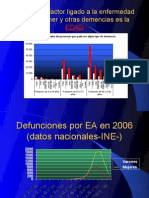
También podría gustarte
- Triptico El Sistema NerviosoDocumento2 páginasTriptico El Sistema NerviosoFoto Estudio Digifot89% (9)
- Corea de HuntingtonDocumento2 páginasCorea de HuntingtonRuben PelaezAún no hay calificaciones
- Agentes Carcinogenicos y Protooncogenes PDFDocumento52 páginasAgentes Carcinogenicos y Protooncogenes PDFCarlos Daniel Torres AvilézAún no hay calificaciones
- Caso Clínico RenalDocumento2 páginasCaso Clínico RenalTracyQuiñonesTafurAún no hay calificaciones
- Fisiologia y Anatomia RenalDocumento47 páginasFisiologia y Anatomia RenalCarlos Daniel Torres AvilézAún no hay calificaciones
- Tomas de Aquino - JusticiaDocumento7 páginasTomas de Aquino - JusticiaCarlos Daniel Torres AvilézAún no hay calificaciones
- I-6. Anticuerpos QuiméricosDocumento11 páginasI-6. Anticuerpos QuiméricosSergiollafeAún no hay calificaciones
- Infografía de Proceso Rompecabezas Sencillo ColoridoDocumento1 páginaInfografía de Proceso Rompecabezas Sencillo ColoridoRubén Rojas BarreraAún no hay calificaciones
- Neuroimagen en Las Demencias PDFDocumento34 páginasNeuroimagen en Las Demencias PDFfreelosophy100% (1)
- G - Prevención de Los Principales Trastornos Mentales de Tipo OrgánicoDocumento10 páginasG - Prevención de Los Principales Trastornos Mentales de Tipo OrgánicoJohn BattleAún no hay calificaciones
- 14 Guia para Medico Sobre El Manejo de La Enfermedad de Huntington.Documento125 páginas14 Guia para Medico Sobre El Manejo de La Enfermedad de Huntington.chabeliAún no hay calificaciones
- F00 Demencia en La Enfermedad de AlzheimerDocumento9 páginasF00 Demencia en La Enfermedad de AlzheimerFabio GonzalezAún no hay calificaciones
- Corea HuntingtonDocumento5 páginasCorea HuntingtonMelany MatusAún no hay calificaciones
- Resume N NeuroDocumento98 páginasResume N NeuroValentina Fernandez ZuletaAún no hay calificaciones
- Involucion Psicomotriz PDFDocumento5 páginasInvolucion Psicomotriz PDFcesarAún no hay calificaciones
- Introducción A Las DemenciasDocumento76 páginasIntroducción A Las Demenciasmatezai100% (2)
- Enfermedades DegenerativasDocumento10 páginasEnfermedades DegenerativasAnalia Montoro100% (1)
- Genetica 1Documento10 páginasGenetica 1Zilary L. Carrasco SeminarioAún no hay calificaciones
- Cuadro Comparativo de Los Transtornos NeuropsicologiaDocumento33 páginasCuadro Comparativo de Los Transtornos NeuropsicologiaRoberto MontesAún no hay calificaciones
- Feria410 01 Ganglios Basales Normal y Sus LesionesDocumento20 páginasFeria410 01 Ganglios Basales Normal y Sus LesionesLorena LazoAún no hay calificaciones
- Quienes Somos (Los Desafíos A La Identidad Estadounidense) - Hugo Gaston SarnoDocumento10 páginasQuienes Somos (Los Desafíos A La Identidad Estadounidense) - Hugo Gaston SarnoCiencia Politica Uahc100% (2)
- Enfermedad de HuntingtonDocumento9 páginasEnfermedad de HuntingtonRogerGarciaAún no hay calificaciones
- El Determinismo GenéticoDocumento7 páginasEl Determinismo GenéticoVladimir VlackAún no hay calificaciones
- Parálisis Cordal (Recurrente y Superior) : EtiologíaDocumento19 páginasParálisis Cordal (Recurrente y Superior) : EtiologíaRomi Belén Cabrera VargasAún no hay calificaciones
- La Terapia Génica en La Enfermedad de Huntington: Una Visión GlobalDocumento1 páginaLa Terapia Génica en La Enfermedad de Huntington: Una Visión GlobalJesus Andrew Valenzuela FigueroaAún no hay calificaciones
- Tabla SindromesDocumento13 páginasTabla SindromesDariana MillanAún no hay calificaciones
- Cap1 Enfermedades Neurodegenerativas Por ProteopatíasDocumento12 páginasCap1 Enfermedades Neurodegenerativas Por ProteopatíasIsabel M RodríguezAún no hay calificaciones
- PAEDocumento23 páginasPAEMariaJoseCarrion0% (1)
- Cuestionario Enfermedades Geneticas.Documento15 páginasCuestionario Enfermedades Geneticas.yomaira malquinAún no hay calificaciones
- Tarea FisiopatologíaDocumento2 páginasTarea FisiopatologíafifeeeAún no hay calificaciones
- Tema 20. Trastornos Del MovimientoDocumento24 páginasTema 20. Trastornos Del MovimientoRenato Vilas BoasAún no hay calificaciones
- Nucleos de La BaseDocumento9 páginasNucleos de La BaseAlexito Elunico EnamoradodealisonAún no hay calificaciones
- Ataxia CerebelosaDocumento46 páginasAtaxia Cerebelosa018200998gAún no hay calificaciones
- Enfermedades NeurodegenerativaDocumento49 páginasEnfermedades NeurodegenerativaThays LopezAún no hay calificaciones