También podría gustarte
- Casos de estudio de termodinámica: Solución mediante el uso de ASPENHYSYSDe EverandCasos de estudio de termodinámica: Solución mediante el uso de ASPENHYSYSCalificación: 4.5 de 5 estrellas4.5/5 (7)
- Robert Merton Teoria y Estructura SocialDocumento4 páginasRobert Merton Teoria y Estructura SocialJulian Escobar Camacho100% (1)
- Informe Leche de Magnesia Con IndiceDocumento9 páginasInforme Leche de Magnesia Con IndiceE Moisés Cochas Escandón100% (2)
- Informe Leche de Magnesia Con IndiceDocumento9 páginasInforme Leche de Magnesia Con IndiceE Moisés Cochas Escandón100% (2)
- Reacciones Químicas y Balanceo de Ecuaciones QuímicasDocumento16 páginasReacciones Químicas y Balanceo de Ecuaciones QuímicasOrly TrianaAún no hay calificaciones
- Diseno Instalac Electricas IEC 60364 6Documento477 páginasDiseno Instalac Electricas IEC 60364 6Leo SanclementeAún no hay calificaciones
- Diseno Instalac Electricas IEC 60364 6Documento477 páginasDiseno Instalac Electricas IEC 60364 6Leo SanclementeAún no hay calificaciones
- 2 ESO REACCIONES QUÍMICAS - Ajuste PDFDocumento5 páginas2 ESO REACCIONES QUÍMICAS - Ajuste PDFJorge ZapicoAún no hay calificaciones
- Reacciones QuimicasDocumento38 páginasReacciones QuimicasFRANCISCA IGNACIA ANTONIA CARVAJAL GUEVARAAún no hay calificaciones
- Balanceo de Ecuaciones QuímicasDocumento9 páginasBalanceo de Ecuaciones Químicasxavig10Aún no hay calificaciones
- Sesion 7-8Documento29 páginasSesion 7-8Byron Pesantez50% (4)
- Catalogo 2021 MONOPOLDocumento90 páginasCatalogo 2021 MONOPOLJosé Luis Vertiz50% (2)
- Acetilacion de Aminas Alifaticas y Aromaticas EstuDocumento6 páginasAcetilacion de Aminas Alifaticas y Aromaticas EstuOscar CardonaAún no hay calificaciones
- Reacciones QuimicasDocumento14 páginasReacciones Quimicascbtis24Aún no hay calificaciones
- Clase+27 Estequiometría+2Documento22 páginasClase+27 Estequiometría+2Maria Alejandra Acosta SáezAún no hay calificaciones
- Ortega DeterminacindeAlyMgenanticidopublicadoDocumento12 páginasOrtega DeterminacindeAlyMgenanticidopublicadojorgeAún no hay calificaciones
- 53874825523838696502020-Clase 3 Formula-EstequiometriaDocumento56 páginas53874825523838696502020-Clase 3 Formula-EstequiometriaSofia AssanAún no hay calificaciones
- 9 - Estequiometría CEIDocumento18 páginas9 - Estequiometría CEIManuel LabraAún no hay calificaciones
- Reactivo LimitanteDocumento18 páginasReactivo LimitanteKarloz Hurtado100% (1)
- Balanceo de Ecuaciones Químicas vs2Documento28 páginasBalanceo de Ecuaciones Químicas vs2rafaelAún no hay calificaciones
- Previo de Laboratorio 5Documento4 páginasPrevio de Laboratorio 5abigail.cruzAún no hay calificaciones
- CURSO DE FISICOQUIMICA II Unidad I Lectura BasicaDocumento57 páginasCURSO DE FISICOQUIMICA II Unidad I Lectura BasicaMiguel Angel Medina CastellanosAún no hay calificaciones
- Alfonso Espinoza Control5Documento6 páginasAlfonso Espinoza Control5aeboladosAún no hay calificaciones
- 5.conceptos de Balance de Mateira en Sistemas - BalanceDocumento46 páginas5.conceptos de Balance de Mateira en Sistemas - Balanceyaquelin.rivera1Aún no hay calificaciones
- Cuadernillo Cuarto A EJE 1 Reacciones y SolucionesDocumento74 páginasCuadernillo Cuarto A EJE 1 Reacciones y SolucionesCarina KriedelAún no hay calificaciones
- Tema4 2esoDocumento3 páginasTema4 2esoMortal ResiduoAún no hay calificaciones
- QSARDocumento28 páginasQSARHoremy Gonzalez HerreraAún no hay calificaciones
- Nivelación Decimo 2018 RefuerzoDocumento4 páginasNivelación Decimo 2018 RefuerzoLedover Ledover LedoverAún no hay calificaciones
- Diapositivas Estequiometria 1Documento15 páginasDiapositivas Estequiometria 1LISSETTE BELTRÁNAún no hay calificaciones
- Reacciones Químicas (Reparado) 1Documento9 páginasReacciones Químicas (Reparado) 1Percil AFAún no hay calificaciones
- Quimica Guía Sumativa N°2 Primer NivelDocumento5 páginasQuimica Guía Sumativa N°2 Primer NivelPaulina VeasAún no hay calificaciones
- Introducción A La EstequiometríaDocumento36 páginasIntroducción A La EstequiometríaEsteban Gabriel SaldiviaAún no hay calificaciones
- Química 2do ParcialDocumento144 páginasQuímica 2do ParcialManu Villalon TellezAún no hay calificaciones
- Quimica Guía Sumativa N°2 Primer NivelDocumento5 páginasQuimica Guía Sumativa N°2 Primer NivelPaulina VeasAún no hay calificaciones
- ESTEQUIOMETRIADocumento10 páginasESTEQUIOMETRIAEliezer GomezAún no hay calificaciones
- Informe Org - Corregido2Documento26 páginasInforme Org - Corregido2Martín SandobalAún no hay calificaciones
- Reacciones Químicas 02Documento14 páginasReacciones Químicas 02Mariana JiménezAún no hay calificaciones
- Guia de Ecuaciones Qui 1mDocumento4 páginasGuia de Ecuaciones Qui 1mXime Lucero JaraAún no hay calificaciones
- Oxidorreducción y AcidezDocumento27 páginasOxidorreducción y AcidezWILFREDO ROMAN PAUCARAún no hay calificaciones
- Ecuaciones QuimicasDocumento24 páginasEcuaciones QuimicasCarlos Fernando Pereira StraubeAún no hay calificaciones
- Complejos Coloridos FeDocumento3 páginasComplejos Coloridos FeGabriel VázquezAún no hay calificaciones
- Informe Determinaciones de CalcioDocumento20 páginasInforme Determinaciones de CalcioJohan RiveraAún no hay calificaciones
- Span EquationsDocumento78 páginasSpan EquationsPau AguilarAún no hay calificaciones
- 3er EXA - QUIMICA 8 - 11Documento12 páginas3er EXA - QUIMICA 8 - 11Brayan Villavicencio TovarAún no hay calificaciones
- Practica 4 Gas IdealDocumento8 páginasPractica 4 Gas IdealDaniel BVAún no hay calificaciones
- Reacciones Químicas Mariana JimenezDocumento13 páginasReacciones Químicas Mariana JimenezMariana JiménezAún no hay calificaciones
- Guias Laboratorio BioquímicaDocumento38 páginasGuias Laboratorio BioquímicaDaniel Felipe FajardoAún no hay calificaciones
- Proyecto Individual Quimica II Primer ParcialDocumento151 páginasProyecto Individual Quimica II Primer ParcialAxel Omar Moreno ValdezAún no hay calificaciones
- Pasos para Calcular Las Ecuaciones QuimicasDocumento2 páginasPasos para Calcular Las Ecuaciones QuimicasJose GutierresAún no hay calificaciones
- Quimica Pau ResumenDocumento2 páginasQuimica Pau Resumenjavibm10Aún no hay calificaciones
- Semana 5 Reacciones Químicas - BalanceoDocumento37 páginasSemana 5 Reacciones Químicas - BalanceoAshly MLAún no hay calificaciones
- Reacciones Quimicas USMPDocumento64 páginasReacciones Quimicas USMPLeslie Rosado MerinoAún no hay calificaciones
- Reacciones Químicas 1º MedioDocumento34 páginasReacciones Químicas 1º MedioFRANCISCA IGNACIA ANTONIA CARVAJAL GUEVARAAún no hay calificaciones
- Aritmetica Química Caso de Estudio Grupo 2Documento12 páginasAritmetica Química Caso de Estudio Grupo 2Giuliana Seña ZubietaAún no hay calificaciones
- Soluciones IDocumento47 páginasSoluciones ISebastián RoqueAún no hay calificaciones
- Clase 08 Termodinamica de Soluciones 1Documento47 páginasClase 08 Termodinamica de Soluciones 1Francisco Javier Alarcón GarridoAún no hay calificaciones
- Guiadeejercicios20171sDocumento12 páginasGuiadeejercicios20171sEsteban GomezAún no hay calificaciones
- Química - La - Reacción - QuímicaDocumento26 páginasQuímica - La - Reacción - QuímicaMarcelo LatojaAún no hay calificaciones
- EstequiometriaDocumento10 páginasEstequiometriadario tonatoAún no hay calificaciones
- Documento de ReiDocumento1 páginaDocumento de ReipandamilovAún no hay calificaciones
- Compendio 2Documento18 páginasCompendio 2Pablo Andrés Morales LeytonAún no hay calificaciones
- P-07-Reacciones Redox (1) Reacciones RedoxDocumento10 páginasP-07-Reacciones Redox (1) Reacciones RedoxBRIGITH STHEFANIA BENITES GARCIAAún no hay calificaciones
- Estequiometria 2Documento40 páginasEstequiometria 2Andres OrdoñezAún no hay calificaciones
- 11 - EstequiometríaDocumento8 páginas11 - Estequiometríawilver Rodriguez avendañoAún no hay calificaciones
- ESTEQUIOMETRIADocumento10 páginasESTEQUIOMETRIAMayanin Arellano FigueroaAún no hay calificaciones
- Aspectos Conceptuales Clase N°16: "Se Define La Química Orgánica Como La Química de Los Compuestos de Carbono."Documento9 páginasAspectos Conceptuales Clase N°16: "Se Define La Química Orgánica Como La Química de Los Compuestos de Carbono."Aracelly JaraAún no hay calificaciones
- Aa 223 Química II e F GDocumento5 páginasAa 223 Química II e F GMATEMÁTICAS UNIAún no hay calificaciones
- Almidon ModificadoDocumento101 páginasAlmidon ModificadoalexandraAún no hay calificaciones
- Procedimiento Trabajo Seguro Con Sustancias PeligrosasDocumento7 páginasProcedimiento Trabajo Seguro Con Sustancias PeligrosasDavid Bedoya MolinaAún no hay calificaciones
- 1672 SPDocumento6 páginas1672 SPE Moisés Cochas EscandónAún no hay calificaciones
- SDB 9370 Es EsDocumento16 páginasSDB 9370 Es EsKelvin EfrainAún no hay calificaciones
- Script Héroes StarMaker AstralisTDocumento2 páginasScript Héroes StarMaker AstralisTE Moisés Cochas EscandónAún no hay calificaciones
- Práctica 1Documento8 páginasPráctica 1E Moisés Cochas EscandónAún no hay calificaciones
- Labo 5 Falta Marco TeoricoDocumento8 páginasLabo 5 Falta Marco TeoricoE Moisés Cochas EscandónAún no hay calificaciones
- Descripción de La Plantilla Del Registro de Los InteresadosDocumento1 páginaDescripción de La Plantilla Del Registro de Los InteresadosE Moisés Cochas EscandónAún no hay calificaciones
- La Triple Restricción y Su ImportanciaDocumento3 páginasLa Triple Restricción y Su ImportanciaE Moisés Cochas EscandónAún no hay calificaciones
- Obedp 2004 BDocumento5 páginasObedp 2004 BE Moisés Cochas EscandónAún no hay calificaciones
- Puertas LogicasDocumento14 páginasPuertas Logicasmglq100% (1)
- SpeechDocumento1 páginaSpeechE Moisés Cochas EscandónAún no hay calificaciones
- EDT Informacion BasicaDocumento7 páginasEDT Informacion BasicaE Moisés Cochas EscandónAún no hay calificaciones
- MaterialeslaboratorioDocumento44 páginasMaterialeslaboratorioJorge Raymundo MelendezAún no hay calificaciones
- Casio News III - Combinatoria y Probabilidad Con La Calculadora ClasswizDocumento2 páginasCasio News III - Combinatoria y Probabilidad Con La Calculadora ClasswizLeonardo AaronAún no hay calificaciones
- La Violencia Contra La MujerDocumento1 páginaLa Violencia Contra La MujerE Moisés Cochas EscandónAún no hay calificaciones
- Argumento y ContraargumentDocumento2 páginasArgumento y ContraargumentE Moisés Cochas Escandón100% (1)
- ARTES PlasticasDocumento4 páginasARTES PlasticasE Moisés Cochas EscandónAún no hay calificaciones
- GRAFENODocumento27 páginasGRAFENOMarcelo CenturyAún no hay calificaciones
- Test Gestión de La IntegraciónDocumento5 páginasTest Gestión de La IntegraciónE Moisés Cochas EscandónAún no hay calificaciones
- El Arte de Bailar KpopDocumento2 páginasEl Arte de Bailar KpopE Moisés Cochas EscandónAún no hay calificaciones
- En Qué Consiste El Filamento PlaDocumento10 páginasEn Qué Consiste El Filamento PlaE Moisés Cochas EscandónAún no hay calificaciones
- Dinamicas de IntegraciónDocumento1 páginaDinamicas de IntegraciónE Moisés Cochas EscandónAún no hay calificaciones
- Guía de Entrenamiento GP FUNDAMENTOSDocumento15 páginasGuía de Entrenamiento GP FUNDAMENTOSrenzoAún no hay calificaciones
- Densidad de LiquidosDocumento12 páginasDensidad de LiquidosE Moisés Cochas EscandónAún no hay calificaciones
- Soluciones - 2010 - 2010 09 28 290Documento13 páginasSoluciones - 2010 - 2010 09 28 290Gabriel BecerraAún no hay calificaciones
- Muestreo y DistribucionesDocumento76 páginasMuestreo y DistribucionesYoselin Dayana Banegas HumireAún no hay calificaciones
- Taller Fork HilosDocumento6 páginasTaller Fork HilosMountain ReAún no hay calificaciones
- LACCEI 2020 - Gonzalez - Aguilar - Huaco PDFDocumento6 páginasLACCEI 2020 - Gonzalez - Aguilar - Huaco PDFElio VidalAún no hay calificaciones
- Ciencias Nat - Guia 2 - 10 y 11 ListaDocumento8 páginasCiencias Nat - Guia 2 - 10 y 11 Listaddanovis0629Aún no hay calificaciones
- Dips 5.1Documento3 páginasDips 5.1Juan Pablo Sanchez MelgarejoAún no hay calificaciones
- Ma470 Cuaderno de Trabajo Marzo 202301Documento219 páginasMa470 Cuaderno de Trabajo Marzo 202301Fiorella Alejandra Cámara LuyoAún no hay calificaciones
- UNIDAD 1 Respuesta en FrecuenciaDocumento79 páginasUNIDAD 1 Respuesta en FrecuenciaVOLTA PROAún no hay calificaciones
- Nivel 5Documento2 páginasNivel 5Dominga RamónAún no hay calificaciones
- Ficha Tecnica Calvente Guindalera 2012Documento1 páginaFicha Tecnica Calvente Guindalera 2012kissjm85Aún no hay calificaciones
- Wilman Leonel Ureña Castro - Asignacion de La Semana Ii-CartografiaDocumento9 páginasWilman Leonel Ureña Castro - Asignacion de La Semana Ii-CartografiaWilman UreñaAún no hay calificaciones
- Criptoanalisis Por KasiskiDocumento8 páginasCriptoanalisis Por KasiskiJvlio Cesar Calleja MorenoAún no hay calificaciones
- 3ero - 1 - MatematicaDocumento17 páginas3ero - 1 - MatematicaEDISON PALMAAún no hay calificaciones
- Pa Tds Parkett Classic-3060 Es 20210401Documento5 páginasPa Tds Parkett Classic-3060 Es 20210401Claudia Escobar TejedaAún no hay calificaciones
- ContenidoDocumento5 páginasContenidofelipe paredesAún no hay calificaciones
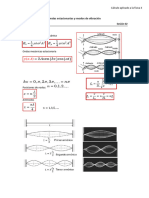
- S Sem6 Ses2 Modos Vibracion-1Documento4 páginasS Sem6 Ses2 Modos Vibracion-1santosml0408Aún no hay calificaciones
- Jaime Segarra El Excitante Camino Al Exito Segunda Edicion GratuitoDocumento7 páginasJaime Segarra El Excitante Camino Al Exito Segunda Edicion Gratuitoerick_abreuvAún no hay calificaciones
- Refrigeración DomesticaDocumento7 páginasRefrigeración DomesticajuniorAún no hay calificaciones
- Mec 2240 Res Aux Sem - 2-2019Documento7 páginasMec 2240 Res Aux Sem - 2-2019Reyna García ChoqueAún no hay calificaciones
- Articulo de Tesis Sistema de Inyección de Pentano para La RMCP en El Yacimiento Bachaquero-01Documento28 páginasArticulo de Tesis Sistema de Inyección de Pentano para La RMCP en El Yacimiento Bachaquero-01gleidyAún no hay calificaciones
- Informe CentroDocumento10 páginasInforme CentroCristhian CardenasAún no hay calificaciones
- Tarea 7Documento14 páginasTarea 7EdgarAún no hay calificaciones
- TP Laboratorio QuimicaDocumento5 páginasTP Laboratorio QuimicaJavier R Hernández100% (2)
- Funciones de Excel Con Sus Ejemplos y AplicacionesDocumento8 páginasFunciones de Excel Con Sus Ejemplos y AplicacionesAnonymous KjWvx8Z4cAún no hay calificaciones
- Analisis Bioclimatico TunjaDocumento4 páginasAnalisis Bioclimatico TunjaeddysayagoAún no hay calificaciones
- 04 - Aplicación Al Diapasón - Nivel 2 PDFDocumento2 páginas04 - Aplicación Al Diapasón - Nivel 2 PDFJoseEscobedoAún no hay calificaciones