También podría gustarte
- Tutorial ChemsketchDocumento3 páginasTutorial ChemsketchXHIARA233Aún no hay calificaciones
- VMD 16 17 Guion4Documento10 páginasVMD 16 17 Guion4MJAún no hay calificaciones
- VMD IntroduccionDocumento12 páginasVMD IntroduccionMilton Paredes AvalosAún no hay calificaciones
- Introduccion Al Programa AMOS 5Documento5 páginasIntroduccion Al Programa AMOS 5jorjisalasAún no hay calificaciones
- AUTOCAD CalibreDocumento3 páginasAUTOCAD CalibreCeleste CmAún no hay calificaciones
- Introducción Al Uso de RasMolDocumento14 páginasIntroducción Al Uso de RasMolAixingruAún no hay calificaciones
- Semana 5 Investigación Sobre Cytoscape 3.6Documento26 páginasSemana 5 Investigación Sobre Cytoscape 3.6EdgarDanielAún no hay calificaciones
- 01 - Building A Geological ModelDocumento34 páginas01 - Building A Geological ModelAlfredo MontalvoAún no hay calificaciones
- 04 Introduccion AMOSDocumento17 páginas04 Introduccion AMOSNoyka Lara100% (1)
- Tutorial Ram ElementsDocumento5 páginasTutorial Ram ElementsmariopelotaAún no hay calificaciones
- Apuntes S3Documento65 páginasApuntes S3Cecilia E. GarciaAún no hay calificaciones
- Tutorial JamoviDocumento25 páginasTutorial JamoviAlber AAGAún no hay calificaciones
- VMD Introduccion PDFDocumento12 páginasVMD Introduccion PDFMilton Paredes AvalosAún no hay calificaciones
- Guia de Actividades y Rubrica de Evaluacion - Unidad 2 - Fase 3 - Distribucion y ProbabilidadDocumento11 páginasGuia de Actividades y Rubrica de Evaluacion - Unidad 2 - Fase 3 - Distribucion y ProbabilidadCESARAún no hay calificaciones
- Composición de Bandas ArcGISDocumento3 páginasComposición de Bandas ArcGISMelquisedec WilchezAún no hay calificaciones
- Manual Uso Rapido Node XLDocumento12 páginasManual Uso Rapido Node XLAnahí reyesAún no hay calificaciones
- Manual Prac QcadDocumento38 páginasManual Prac QcadoroxcooAún no hay calificaciones
- Manual Laser5.3Documento37 páginasManual Laser5.3Juan Asmat100% (1)
- Ejercicio 2Documento8 páginasEjercicio 2Javier López MarínAún no hay calificaciones
- Analisis de Fragmentacion Granulometria en Voladura de Rocas Con ImageJ Romel Villanueva Iiming PDFDocumento14 páginasAnalisis de Fragmentacion Granulometria en Voladura de Rocas Con ImageJ Romel Villanueva Iiming PDFAnonymous UXEtI80kAún no hay calificaciones
- CadstdDocumento23 páginasCadstdEdgar Rodriguez MartinezAún no hay calificaciones
- Instrucciones de Ejercicio Taller de CatastroDocumento7 páginasInstrucciones de Ejercicio Taller de CatastroCortez AliciaAún no hay calificaciones
- Practicas de Moviles 2023Documento39 páginasPracticas de Moviles 2023Manuel GonzalezAún no hay calificaciones
- Tutorial EagleDocumento14 páginasTutorial EaglesoymoeAún no hay calificaciones
- Guia de Actividades y Rubrica de Evaluacion - Unidad 2 - Fase 3 - Distribucion y ProbabilidadDocumento12 páginasGuia de Actividades y Rubrica de Evaluacion - Unidad 2 - Fase 3 - Distribucion y Probabilidadyenidfer parraAún no hay calificaciones
- Guia de Usuario de Pivlab2Documento5 páginasGuia de Usuario de Pivlab2JC2514Aún no hay calificaciones
- Taller1 - Hector Luis GalindezDocumento36 páginasTaller1 - Hector Luis GalindezHECTOR LUIS GALINDEZ BURBANOAún no hay calificaciones
- Tutorial DatamineDocumento13 páginasTutorial DatamineCristian EddlerAún no hay calificaciones
- TP 5.1 Clasificación Supervisada de ImágenesDocumento22 páginasTP 5.1 Clasificación Supervisada de ImágenesMatías QuintanaAún no hay calificaciones
- ModelamientoDocumento5 páginasModelamientoEspiritu Espiritu HiberAún no hay calificaciones
- Sensores Landsat 5 Segunda PracticaDocumento8 páginasSensores Landsat 5 Segunda PracticaFRANCISCO TOMAS GALLEGOS RANILLAAún no hay calificaciones
- Manual PSPPDocumento25 páginasManual PSPPVictor Angel ChanAún no hay calificaciones
- PRÁCTICAS Con QCAD (3º ESO)Documento39 páginasPRÁCTICAS Con QCAD (3º ESO)Pablo López100% (2)
- Libro1 (Recuperado Automáticamente) (Recuperado Automáticamente) (Recuperado Automáticamente)Documento84 páginasLibro1 (Recuperado Automáticamente) (Recuperado Automáticamente) (Recuperado Automáticamente)Libardo MorenoAún no hay calificaciones
- Tubo Laminar PDFDocumento15 páginasTubo Laminar PDFIlae RuizAún no hay calificaciones
- Guía Ejercicio de Simul 8Documento7 páginasGuía Ejercicio de Simul 8John OrtizAún no hay calificaciones
- Guia - SRTM MODELOS DE ELEVACION DIGITALDocumento12 páginasGuia - SRTM MODELOS DE ELEVACION DIGITALariane jimenezAún no hay calificaciones
- PHASE2 Tutorial Inicio RápidoDocumento24 páginasPHASE2 Tutorial Inicio RápidoPatricioAntonioDonosoAún no hay calificaciones
- Unidad 3 Autocad 2DDocumento16 páginasUnidad 3 Autocad 2Djose castroAún no hay calificaciones
- PasosDocumento20 páginasPasosIrene Arribas EstebanAún no hay calificaciones
- Proceso de Construccion ArmaduraDocumento18 páginasProceso de Construccion ArmaduraHeriberto Barragán MuñozAún no hay calificaciones
- Analisis de Fragmentación - Granulometria en Voladura de Rocas Con ImageJ - Romel Villanueva - IimingDocumento14 páginasAnalisis de Fragmentación - Granulometria en Voladura de Rocas Con ImageJ - Romel Villanueva - IimingRomel Villanueva60% (5)
- Guia de Actividades Clase 1Documento6 páginasGuia de Actividades Clase 1Ezequiel LuppiAún no hay calificaciones
- VALDIVIADocumento23 páginasVALDIVIAanyAún no hay calificaciones
- Acad2002 Leccion7Documento14 páginasAcad2002 Leccion7MiguelAún no hay calificaciones
- Autocad Escalar Impresion1Documento10 páginasAutocad Escalar Impresion1RAMIRINGARAún no hay calificaciones
- Parte 1 PDFDocumento39 páginasParte 1 PDFAnonymous znPzQTaYoAún no hay calificaciones
- QGISDocumento18 páginasQGISOmar L ApazaAún no hay calificaciones
- Tutorial Algebra de BandasDocumento4 páginasTutorial Algebra de BandasEmilio Patané SpataroAún no hay calificaciones
- Estadistica - Unidad 2 - Fase 3 - Distribucion y ProbabilidadDocumento10 páginasEstadistica - Unidad 2 - Fase 3 - Distribucion y ProbabilidadMaicol Vargas100% (1)
- Como Utilizar Meshlab y Netfabb para Arreglar Tu Objeto 3D - Impresoras3dDocumento3 páginasComo Utilizar Meshlab y Netfabb para Arreglar Tu Objeto 3D - Impresoras3dingeomar2001Aún no hay calificaciones
- Fase 4Documento6 páginasFase 4Alonso Javier Suarez AmayaAún no hay calificaciones
- Trabajo Semanal15 - 08 - 2021 Preguntas 3 y 4 RevisarDocumento5 páginasTrabajo Semanal15 - 08 - 2021 Preguntas 3 y 4 RevisarRAUL WASHINGTON MESCCO PUMAAún no hay calificaciones
- Primer Informe MT420 2020-2Documento39 páginasPrimer Informe MT420 2020-2samuelAún no hay calificaciones
- Apuntes EtalonajeDocumento3 páginasApuntes EtalonajeGabriel San PabloAún no hay calificaciones
- Módulo de cinemática DMU de Catia V5De EverandMódulo de cinemática DMU de Catia V5Calificación: 5 de 5 estrellas5/5 (1)
- Matplotlib, Introducción a la Visualización 2D, Parte IDe EverandMatplotlib, Introducción a la Visualización 2D, Parte IAún no hay calificaciones
- Introducción al modelado matemático con MatLabDe EverandIntroducción al modelado matemático con MatLabAún no hay calificaciones
- Editora de gráficos ráster: Transformando realidades visuales: dominio de los editores de gráficos rasterizados en visión por computadoraDe EverandEditora de gráficos ráster: Transformando realidades visuales: dominio de los editores de gráficos rasterizados en visión por computadoraAún no hay calificaciones
- Teorico de Hidrocarburos Aromaticos Polinucleares 2017Documento35 páginasTeorico de Hidrocarburos Aromaticos Polinucleares 2017whiyeAún no hay calificaciones
- Titanic (Ing) PDFDocumento400 páginasTitanic (Ing) PDFwhiyeAún no hay calificaciones
- Titanic (Ing) PDFDocumento400 páginasTitanic (Ing) PDFwhiyeAún no hay calificaciones
- Practica 2Documento6 páginasPractica 2whiyeAún no hay calificaciones
- Analisis Matemático-Puntos CriticosDocumento20 páginasAnalisis Matemático-Puntos CriticoswhiyeAún no hay calificaciones
- Alberts Cap 20 Celulas Germinales y FecundacionDocumento22 páginasAlberts Cap 20 Celulas Germinales y FecundacionwhiyeAún no hay calificaciones
- Movimimo 32Documento36 páginasMovimimo 32Camila TellecheaAún no hay calificaciones
- ANALISIS DE PRECIOS UNITARIOS-ing NavarroDocumento85 páginasANALISIS DE PRECIOS UNITARIOS-ing NavarroMax Lenin Ticona CondoriAún no hay calificaciones
- Argumentacion Gema MarcillaDocumento5 páginasArgumentacion Gema MarcillaMaríaCandelariaQuispePonceAún no hay calificaciones
- CURSO ODOO 17 - CLASGURU - BoDocumento7 páginasCURSO ODOO 17 - CLASGURU - BoChristian Emanuel Paredes AlanesAún no hay calificaciones
- Pae Completo Ya ListoDocumento26 páginasPae Completo Ya Listoyane50% (2)
- Lectura Poética Aristóteles 2Documento11 páginasLectura Poética Aristóteles 2LIZETH KATERINE CANO ESCOBARAún no hay calificaciones
- Instructivo para Pagos de CesantíasDocumento13 páginasInstructivo para Pagos de CesantíasDora Maria Cobos VillarragaAún no hay calificaciones
- Clase Comercio Internacional Dia 20 de AbrilDocumento2 páginasClase Comercio Internacional Dia 20 de AbrilLizett Kokalli PichisAún no hay calificaciones
- 03 Propiedades de Las PotenciasDocumento21 páginas03 Propiedades de Las PotenciasDavid Esteban Quezada MieresAún no hay calificaciones
- Mpu Panama Pppea 0 PoliticaDocumento21 páginasMpu Panama Pppea 0 PoliticaMarilyn SugastyAún no hay calificaciones
- Vuelta Al Cole.Documento5 páginasVuelta Al Cole.NATALIABCAún no hay calificaciones
- Metodologia Youdon-DeMarcoDocumento3 páginasMetodologia Youdon-DeMarcoFernando ScAún no hay calificaciones
- Ensayo de Partidos PoliticosDocumento4 páginasEnsayo de Partidos Politicosdaniel50% (4)
- Texto Resistencia Corredores-LeibarDocumento248 páginasTexto Resistencia Corredores-LeibarOskar VillamarAún no hay calificaciones
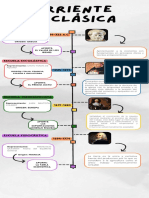
- Linea de Tiempo Corriente PreclásicaDocumento1 páginaLinea de Tiempo Corriente PreclásicaRosa EstradaAún no hay calificaciones
- Plan de Trabajo Inmunizaciones Huayllasp 2022Documento7 páginasPlan de Trabajo Inmunizaciones Huayllasp 2022P.S Huayllaspanca67% (6)
- Artículo CodependenciaDocumento3 páginasArtículo Codependenciazulema52Aún no hay calificaciones
- Grados de Tubería de Producción J55, N80, C75, P110Documento11 páginasGrados de Tubería de Producción J55, N80, C75, P110ANDREA NICOLE TOLEDO CAZASAún no hay calificaciones
- Resumen de EmelyDocumento16 páginasResumen de EmelyMaximo J. ParedesAún no hay calificaciones
- Estudio de Caso-Gestión de Un Ava Utilizando El Ciclo PhvaDocumento5 páginasEstudio de Caso-Gestión de Un Ava Utilizando El Ciclo PhvaPilar MAún no hay calificaciones
- Chonta CuestionarioDocumento43 páginasChonta CuestionarioCarlos BustamanteAún no hay calificaciones
- DTR1695Documento1 páginaDTR1695Tomas BadilloAún no hay calificaciones
- Curso Coordinacion de Protecciones Electricas en Redes de Distribucion Clase 1.1Documento13 páginasCurso Coordinacion de Protecciones Electricas en Redes de Distribucion Clase 1.1Lalex MoretaAún no hay calificaciones
- Angel Guimera 5 Hospitalet Dossier en Vigor Abril 2022 Renders y FachadaDocumento21 páginasAngel Guimera 5 Hospitalet Dossier en Vigor Abril 2022 Renders y FachadaNélidaAún no hay calificaciones
- La Hija Del Sha PDFDocumento5 páginasLa Hija Del Sha PDFmaxito0229Aún no hay calificaciones
- 67 Pemex SRHRLDocumento7 páginas67 Pemex SRHRLPos Yo YoAún no hay calificaciones
- Aportes Psicologia de La SexualidadDocumento3 páginasAportes Psicologia de La SexualidadJulieth CardenasAún no hay calificaciones
- Fracturas en NiñosDocumento15 páginasFracturas en NiñosKarina HernandezAún no hay calificaciones
- Libro Matemática Álgebra de NúmerosDocumento9 páginasLibro Matemática Álgebra de NúmerosHeider David Salazar100% (1)
- 1964 Vida - Con - El - LamaDocumento215 páginas1964 Vida - Con - El - LamaKathia D KostlichAún no hay calificaciones