También podría gustarte
- Laboratorio PCRDocumento9 páginasLaboratorio PCRWendy Quiroz BerdugoAún no hay calificaciones
- Tecl13 PDFDocumento99 páginasTecl13 PDFThuLokithaMermaAún no hay calificaciones
- PCRDocumento4 páginasPCRpauAún no hay calificaciones
- ResumenDocumento7 páginasResumenBetsy RodriguezAún no hay calificaciones
- CitogenéticaDocumento5 páginasCitogenéticaAriel Rolón0% (1)
- PRIMERA EVALUACIÓN PRÁCTICA DE GENÉTICADocumento1 páginaPRIMERA EVALUACIÓN PRÁCTICA DE GENÉTICAMiguelangel Ayca ChambillaAún no hay calificaciones
- Interpretación de La Reaccion en Cadena de La PolimerasaDocumento7 páginasInterpretación de La Reaccion en Cadena de La Polimerasa.:("*"BLacK BuLLeT"*"):.100% (3)
- Qué es la reacción en cadena de la polimerasa (PCRDocumento70 páginasQué es la reacción en cadena de la polimerasa (PCRNicolas rodriguez0% (1)
- Práctica 5 - 1 - LìpidosDocumento9 páginasPráctica 5 - 1 - LìpidosIvan NuñezAún no hay calificaciones
- Unidad 4 Filosofia de Los HongosDocumento25 páginasUnidad 4 Filosofia de Los Hongosalejandra diazAún no hay calificaciones
- Exam. 11Documento3 páginasExam. 11Helen BravoAún no hay calificaciones
- PCRDocumento43 páginasPCRSam CuocoAún no hay calificaciones
- Cuadro Sinoptico de SNDocumento1 páginaCuadro Sinoptico de SNAlejandra Rodriguez100% (1)
- Examen Tercer Parcial Neuro 1Documento30 páginasExamen Tercer Parcial Neuro 1Andrea Fernanda SánchezAún no hay calificaciones
- Informe 4 Laboratorio BtaDocumento10 páginasInforme 4 Laboratorio BtaGabriela GonzalezAún no hay calificaciones
- Calor en BiofisicaDocumento39 páginasCalor en BiofisicaYobahana Vallejo0% (1)
- Resistencia y La Ecuacion de PoiseuilleDocumento3 páginasResistencia y La Ecuacion de PoiseuilleShantal VegaAún no hay calificaciones
- Tinciones hematológicas convencionalesDocumento3 páginasTinciones hematológicas convencionalesJuan Miguel Caracuel LòpezAún no hay calificaciones
- Fiebre TifoideaDocumento4 páginasFiebre TifoideaWilliam EliezerAún no hay calificaciones
- RM en EpilepsiaDocumento60 páginasRM en EpilepsiaJavier SanchezAún no hay calificaciones
- Bio - ChagasDocumento4 páginasBio - ChagasYazmin L. FlowersAún no hay calificaciones
- Experimiento #2: Antagonismo Fisiológico: "Intensidad Del Efecto"Documento4 páginasExperimiento #2: Antagonismo Fisiológico: "Intensidad Del Efecto"Milagros Vela SalazarAún no hay calificaciones
- Hanes WoolfDocumento8 páginasHanes Woolfcirobv19Aún no hay calificaciones
- Probabilidades - Estrategias - Aldo Trucios Cornejo - 1325210335Documento36 páginasProbabilidades - Estrategias - Aldo Trucios Cornejo - 1325210335Aldo Trucios CornejoAún no hay calificaciones
- Reproduccion AsexualDocumento3 páginasReproduccion AsexualVanesa Muñoz100% (1)
- Tarea QPCRDocumento3 páginasTarea QPCRleonardo manrique100% (1)
- Detecta fragmentos ADN mediante Southern blotDocumento18 páginasDetecta fragmentos ADN mediante Southern blotRocio RubioAún no hay calificaciones
- PCRDocumento7 páginasPCREstefanía AltamiranoAún no hay calificaciones
- Proteoma y Transcriptoma 2014-1Documento67 páginasProteoma y Transcriptoma 2014-1Fernanda MartinezAún no hay calificaciones
- Informe 1 FinalDocumento5 páginasInforme 1 FinalNico ZabalaAún no hay calificaciones
- Informe de Pasantias Patología Medica UAMDocumento6 páginasInforme de Pasantias Patología Medica UAMYhismeldCardenasAún no hay calificaciones
- Semana 1 Diagnosticos de Los Casos ClínicosDocumento13 páginasSemana 1 Diagnosticos de Los Casos ClínicosSteven Flores AijaAún no hay calificaciones
- Caso Clinico Hepatitis CDocumento4 páginasCaso Clinico Hepatitis CKaterine Antequera MartinezAún no hay calificaciones
- LINFOMA DE HODGKIN: CARACTERÍSTICAS, CLASIFICACIÓN E IMPLICACIONESDocumento20 páginasLINFOMA DE HODGKIN: CARACTERÍSTICAS, CLASIFICACIÓN E IMPLICACIONESRosales Cruzalegui K. MikaelAún no hay calificaciones
- Caso Clínico Paro CardiacoDocumento2 páginasCaso Clínico Paro CardiacoEmanuel CFAún no hay calificaciones
- Técnica de PCRDocumento29 páginasTécnica de PCRWendy MayorgaAún no hay calificaciones
- Anticuerpos Antinucleares Clinica DiagnosticaDocumento6 páginasAnticuerpos Antinucleares Clinica Diagnosticaeduardo aponte100% (4)
- Potencial de Membrana de La Célula NerviosaDocumento8 páginasPotencial de Membrana de La Célula NerviosaMATEO YONATHAN CABRERA GAMBOAAún no hay calificaciones
- PCR, PCR Multiplex ExposicionDocumento32 páginasPCR, PCR Multiplex ExposicionGabby Alvarez Oviedo100% (1)
- Albumina en SueroDocumento5 páginasAlbumina en SueroQUIMICO CLINICO WILLIANS SANCHEZ100% (2)
- PÚRPURADocumento15 páginasPÚRPURAJanethSalvadorAún no hay calificaciones
- PCR Transcriptasa ReversaDocumento2 páginasPCR Transcriptasa ReversaLuis S. Montoya100% (3)
- CASO CLÍNICO - Castro - OlenkaDocumento15 páginasCASO CLÍNICO - Castro - OlenkaOlenka CastroAún no hay calificaciones
- 2015 Práctica I GenéticaDocumento7 páginas2015 Práctica I GenéticaMario Orlando Chumioque SanchezAún no hay calificaciones
- Bicompartimental FarmacologiaDocumento20 páginasBicompartimental Farmacologiakyasarin12Aún no hay calificaciones
- Taller 3 - BÚSQUEDA DE SECUENCIAS DE GENES NUCLEARESDocumento16 páginasTaller 3 - BÚSQUEDA DE SECUENCIAS DE GENES NUCLEARESGianella GarcésAún no hay calificaciones
- Importancia de la troponina T cardiaca en el diagnóstico de daño miocárdicoDocumento16 páginasImportancia de la troponina T cardiaca en el diagnóstico de daño miocárdicoAngel Bueno OchoaAún no hay calificaciones
- Dimeros D Analisis de Sangre 2686 Mvi540Documento2 páginasDimeros D Analisis de Sangre 2686 Mvi540thorin8100% (1)
- Técnicas de Hibridación de Los Ácidos NucleicosDocumento95 páginasTécnicas de Hibridación de Los Ácidos NucleicosMaria GomezAún no hay calificaciones
- BiofisicaDocumento2 páginasBiofisicamelissaAún no hay calificaciones
- Cuestiuonerio 2Documento31 páginasCuestiuonerio 2Melanie Castro100% (1)
- Exposicion TermoquimicaDocumento44 páginasExposicion Termoquimicahercam94Aún no hay calificaciones
- Manual de HematologiaDocumento86 páginasManual de Hematologialinda1203Aún no hay calificaciones
- Funcion TubularDocumento46 páginasFuncion TubularRosita Pomalazo PalominoAún no hay calificaciones
- Anatomía y Embriología de Órganos y SistemasDocumento167 páginasAnatomía y Embriología de Órganos y SistemasCristóbal Muñoz Pérez100% (2)
- ADNDocumento3 páginasADNSaid Gabriel JiménezAún no hay calificaciones
- La Reacción en Cadena de La PolimerasaDocumento4 páginasLa Reacción en Cadena de La PolimerasaBlaster X-fixAún no hay calificaciones
- PCRDocumento6 páginasPCRJuliana CorderoAún no hay calificaciones
- Qué Es La PCRDocumento4 páginasQué Es La PCRCamila ParraAún no hay calificaciones
- Guía 9. PCRDocumento3 páginasGuía 9. PCROscar GuarinAún no hay calificaciones
- YamahaXTZ125 Ed58Documento5 páginasYamahaXTZ125 Ed58Juan Manuel Vicencio ArriagadaAún no hay calificaciones
- Data Panel TheoryDocumento10 páginasData Panel TheoryDaniel RomeroAún no hay calificaciones
- Clio TerciosDocumento12 páginasClio TerciosDaniel RomeroAún no hay calificaciones
- Competiti Vi DadDocumento18 páginasCompetiti Vi DadHolger QuispeAún no hay calificaciones
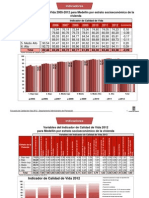
- Indicador de Calidad de Vida 2005-2012Documento6 páginasIndicador de Calidad de Vida 2005-2012Daniel RomeroAún no hay calificaciones
- Como EscribirDocumento2 páginasComo EscribirDaniel RomeroAún no hay calificaciones
- Presentación DanielDocumento46 páginasPresentación DanielDaniel RomeroAún no hay calificaciones
- Seguridad DemocraticaDocumento45 páginasSeguridad DemocraticaDaniel RomeroAún no hay calificaciones
- TeoriadeconsumoDocumento60 páginasTeoriadeconsumozigantoeAún no hay calificaciones
- Seguridad DemocraticaDocumento45 páginasSeguridad DemocraticaDaniel RomeroAún no hay calificaciones
- EconofisicaDocumento8 páginasEconofisicaSanti SlvAún no hay calificaciones
- Arango - Gonzalo-Muerte No Seas MujerDocumento3 páginasArango - Gonzalo-Muerte No Seas MujerEl Flaco Santa RosaAún no hay calificaciones
- Como Entender y Hacer Demostraciones Matematicas PDFDocumento181 páginasComo Entender y Hacer Demostraciones Matematicas PDFJosé María Silvano García Arellano100% (1)
- Carestia de Los Alimentos El Precio Del ExitoDocumento2 páginasCarestia de Los Alimentos El Precio Del ExitoDaniel RomeroAún no hay calificaciones
- Como Entender y Hacer Demostraciones Matematicas PDFDocumento181 páginasComo Entender y Hacer Demostraciones Matematicas PDFJosé María Silvano García Arellano100% (1)
- El Resumen de Un Articulo CientificoDocumento4 páginasEl Resumen de Un Articulo CientificoDaniel RomeroAún no hay calificaciones
- Tutorial DeriveDocumento21 páginasTutorial DeriveRaul SuveAún no hay calificaciones
- Leyes de MendelDocumento2 páginasLeyes de MendelJesúsAún no hay calificaciones
- Nociones de Genética MatematicaDocumento71 páginasNociones de Genética MatematicaAugusto Ǝxcelmes Cutimbo100% (1)
- 2° Medio Etapas Del Ciclo CelularDocumento30 páginas2° Medio Etapas Del Ciclo CelularRayen BeraAún no hay calificaciones
- Proyecto Genoma Humano: Amin El Bouhoutani Aghmir 4ºBDocumento8 páginasProyecto Genoma Humano: Amin El Bouhoutani Aghmir 4ºBAmin El BouhoutaniAún no hay calificaciones
- Trasplante de Células Madre EmbrionariasDocumento24 páginasTrasplante de Células Madre EmbrionariasMARIAM MIYANAY UMERES BRAVO100% (3)
- Síndrome-Expo-Ciclo ViDocumento6 páginasSíndrome-Expo-Ciclo ViAmixclara RoniAún no hay calificaciones
- Genes Y Genomas: Actividad ColaborativaDocumento9 páginasGenes Y Genomas: Actividad ColaborativaUnicornio bonitoAún no hay calificaciones
- Apuntes Bioquimica Udemy Maestro Camach Leanr Ingeniero en BiotecnologíaDocumento2 páginasApuntes Bioquimica Udemy Maestro Camach Leanr Ingeniero en BiotecnologíaHader CruzAún no hay calificaciones
- Lunes, 17 de Mayo de 2021, 16:18 Finalizado Lunes, 17 de Mayo de 2021, 16:47 29 Minutos 10 Segundos 7,00/10,00 0,88 de 1,25 (70%)Documento6 páginasLunes, 17 de Mayo de 2021, 16:18 Finalizado Lunes, 17 de Mayo de 2021, 16:47 29 Minutos 10 Segundos 7,00/10,00 0,88 de 1,25 (70%)Laura HerasmeAún no hay calificaciones
- La herencia genética: características hereditarias y determinación del sexoDocumento3 páginasLa herencia genética: características hereditarias y determinación del sexojean pierre calixto huamánAún no hay calificaciones
- El Adn de La Sanacin Margaret Ruby CH PDFDocumento151 páginasEl Adn de La Sanacin Margaret Ruby CH PDFEdgar HernandezAún no hay calificaciones
- Guía Síntesis de ProteínasDocumento7 páginasGuía Síntesis de ProteínasRomayne MarianaAún no hay calificaciones
- TraducciónDocumento8 páginasTraducciónMaria FernandaAún no hay calificaciones
- Sexo en PlantasDocumento10 páginasSexo en PlantasRubenFrancoAún no hay calificaciones
- LMAcon Anormalidades GeneticasDocumento20 páginasLMAcon Anormalidades GeneticasRudy JumperAún no hay calificaciones
- A17 ProblemasdegeneticaDocumento12 páginasA17 ProblemasdegeneticaJhon Garcia LopezAún no hay calificaciones
- Mejora Genetica AnimalDocumento6 páginasMejora Genetica AnimalPaul Hawkins AlcantaraAún no hay calificaciones
- Mutaciones CromosómicasDocumento22 páginasMutaciones CromosómicasDiego Núñez AlonsoAún no hay calificaciones
- Mapa de Conflictos Éticos en Edición Gen. 2019. CRISPRDocumento3 páginasMapa de Conflictos Éticos en Edición Gen. 2019. CRISPRJuan Jose Eraso OsorioAún no hay calificaciones
- Cariotipo y Mutaciones BiologiaDocumento8 páginasCariotipo y Mutaciones BiologiaLiliana ElizabethAún no hay calificaciones
- Leyes de MendelDocumento6 páginasLeyes de MendelDavid FlorezAún no hay calificaciones
- Punnett Cuadro PDFDocumento3 páginasPunnett Cuadro PDFJuan Sebastian Ruiz RuizAún no hay calificaciones
- Resumen UP2Documento11 páginasResumen UP2SofiaAún no hay calificaciones
- Gregor Mendel, el padre de la genéticaDocumento12 páginasGregor Mendel, el padre de la genéticaYon quispe gomezAún no hay calificaciones
- Grupo 10 Primer Informe BiotecnologiaDocumento19 páginasGrupo 10 Primer Informe BiotecnologiaRUTH ELIZABETH PUMA YAULIAún no hay calificaciones
- Genes Evolucion y ConductaDocumento11 páginasGenes Evolucion y ConductaLauraAún no hay calificaciones
- Pruebas MoleDocumento279 páginasPruebas MoleANGHELO SEBASTHIAN GAVILANEZ DIAZAún no hay calificaciones
- Semana 30 Sesión Lunes 26 Octubre Ciclo Vii 5° Grado Ciencia y TecnologíaDocumento4 páginasSemana 30 Sesión Lunes 26 Octubre Ciclo Vii 5° Grado Ciencia y TecnologíaVictor AzañedoAún no hay calificaciones
- Clase 2. Mitosis y Acidos NucleicosDocumento7 páginasClase 2. Mitosis y Acidos NucleicosRosaynes Romero LoboAún no hay calificaciones
- Transformación de Plantas de Tomate VerdeDocumento12 páginasTransformación de Plantas de Tomate VerdeJorgeCesarCisnerosFigueroaAún no hay calificaciones