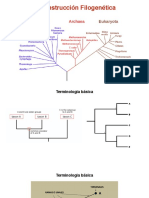
También podría gustarte
- Clase 1.2. Bioinformatica 2023 - 1Documento49 páginasClase 1.2. Bioinformatica 2023 - 1sheila paola correaAún no hay calificaciones
- Analisis de Secuencias I PDFDocumento71 páginasAnalisis de Secuencias I PDFJose Antonio Vargas GonzalezAún no hay calificaciones
- 23 AlineamientosDocumento34 páginas23 AlineamientosfrankAún no hay calificaciones
- 3alineamiento PAREADO de Secuencias2013Documento36 páginas3alineamiento PAREADO de Secuencias2013Orlando SevillanoAún no hay calificaciones
- BioinformáticaDocumento6 páginasBioinformáticaJoseAún no hay calificaciones
- Alineamiento MãºltipleDocumento26 páginasAlineamiento MãºltipleKimberly Fuentes ValdésAún no hay calificaciones
- Unidad1 - AnalisisdeSecuencias - B (3-4)Documento50 páginasUnidad1 - AnalisisdeSecuencias - B (3-4)Micaela AvacaAún no hay calificaciones
- T3 AlineacionsecuenciasDocumento71 páginasT3 AlineacionsecuenciasF CPAún no hay calificaciones
- Conferencia IA-5 Algoritmo GenéticoDocumento27 páginasConferencia IA-5 Algoritmo GenéticoDanielaAún no hay calificaciones
- Unidad1 AnálisisdeSecuencias ADocumento59 páginasUnidad1 AnálisisdeSecuencias AMicaela AvacaAún no hay calificaciones
- Alineamineto de Secuencias-GDocumento68 páginasAlineamineto de Secuencias-GGerardina VandeshourtAún no hay calificaciones
- Bioinformatica TeoriaDocumento65 páginasBioinformatica TeoriaFAUSTO GABRIEL YEPEZ CALDERONAún no hay calificaciones
- Bioinfo 3Documento10 páginasBioinfo 3Cristina TaipeAún no hay calificaciones
- Libro 03 Temab3 21Documento50 páginasLibro 03 Temab3 21Virginia COAún no hay calificaciones
- Teórico 5 - Modelado Estructural de ProteínasDocumento32 páginasTeórico 5 - Modelado Estructural de ProteínasMariana AgustinaAún no hay calificaciones
- Evaluación Continua Virtual PDFDocumento5 páginasEvaluación Continua Virtual PDFKrizz Melgarejo Olortegui100% (1)
- Uso Mtdfreml - YjrDocumento17 páginasUso Mtdfreml - YjrYamileth Jerónimo RomeroAún no hay calificaciones
- Analisis Post Test TELEPRE 1Documento3 páginasAnalisis Post Test TELEPRE 1Constanza Morales CatalanAún no hay calificaciones
- Exposicion Genetic ProgrammingDocumento26 páginasExposicion Genetic ProgrammingCamilo CanoAún no hay calificaciones
- Analisis Semantico PDFDocumento18 páginasAnalisis Semantico PDFStewart RoyceAún no hay calificaciones
- Sesión 21 Regresión Bivariada Los Supuestos 27.10.20 XDocumento37 páginasSesión 21 Regresión Bivariada Los Supuestos 27.10.20 XCristóbal Karle SaavedraAún no hay calificaciones
- Prueba T: NotasDocumento4 páginasPrueba T: NotasJosé Soto CapuchoAún no hay calificaciones
- STATA4 Infer Xid-995144 1Documento26 páginasSTATA4 Infer Xid-995144 1Naomi CRTAún no hay calificaciones
- Caracteres Moleculares 2007Documento54 páginasCaracteres Moleculares 2007Racso Erriuga OdnaboAún no hay calificaciones
- Graficos 2 T StudentDocumento6 páginasGraficos 2 T StudentLex LiterasAún no hay calificaciones
- ProyectoT1 PDFDocumento18 páginasProyectoT1 PDFDaniel LopezAún no hay calificaciones
- ModeloALINEAMIENTO SEQpdfDocumento15 páginasModeloALINEAMIENTO SEQpdfFrancisco FernandezAún no hay calificaciones
- Problemas de Matrices y Determinantes para Quinto de SecundariaDocumento7 páginasProblemas de Matrices y Determinantes para Quinto de SecundariaJacxy FerrerAún no hay calificaciones
- Matrices y DeterminantesDocumento5 páginasMatrices y Determinantesjuan carlosAún no hay calificaciones
- Tema2 Aln Pareados y BLASTDocumento20 páginasTema2 Aln Pareados y BLASTJames ReinaAún no hay calificaciones
- Herramientas InformáticasDocumento20 páginasHerramientas InformáticasLetty EAún no hay calificaciones
- Segundo Parcial - Presencial - Funciones y Estructuras SelectivasDocumento40 páginasSegundo Parcial - Presencial - Funciones y Estructuras Selectivasallan.fernando.reyes.pachecoAún no hay calificaciones
- BioinformáticaDocumento4 páginasBioinformáticaNick Kevin Silva G100% (1)
- 2021 1 Programacion 1 PC1Documento14 páginas2021 1 Programacion 1 PC1Abel Jaime Rivera OlazaAún no hay calificaciones
- 3 - Alineamientos de ParesDocumento73 páginas3 - Alineamientos de ParesAixingruAún no hay calificaciones
- Bioinformatica 4Documento19 páginasBioinformatica 4Santiago ToapantaAún no hay calificaciones
- 4 Blast PDFDocumento75 páginas4 Blast PDFCrist VillarAún no hay calificaciones
- ValidaciónDocumento34 páginasValidaciónOmar MejíaAún no hay calificaciones
- Class 70 Sequence AlignmentDocumento300 páginasClass 70 Sequence AlignmentqatcoAún no hay calificaciones
- MATLAB - FuncionesDocumento100 páginasMATLAB - FuncionesVictor Báez PérezAún no hay calificaciones
- SSD Diapo 4ADocumento20 páginasSSD Diapo 4AMartin Manuel Patiño AbantoAún no hay calificaciones
- Tablas Cruzadas Maria FinalDocumento7 páginasTablas Cruzadas Maria FinalValentina CarrilloAún no hay calificaciones
- KBIT Hoja de RespuestasDocumento9 páginasKBIT Hoja de RespuestasAlejandro Benavides DiagoAún no hay calificaciones
- Analytics PhytonDocumento153 páginasAnalytics PhytonEliana Rocio Salas CasasAún no hay calificaciones
- Examen de BioinformaticaDocumento14 páginasExamen de BioinformaticaJordi Ysmael Camara CabelloAún no hay calificaciones
- Introducción MatlabDocumento17 páginasIntroducción MatlabMatias Villanueva RochefortAún no hay calificaciones
- Teoria DSDocumento6 páginasTeoria DSRaul Alejandro GrassiAún no hay calificaciones
- Clase 01 Procesos InfinitosDocumento9 páginasClase 01 Procesos InfinitosNAOMÍ SALOMÉ PINO SANTANAAún no hay calificaciones
- OUTPUT2CAJADocumento41 páginasOUTPUT2CAJALeila Padilla BenitezAún no hay calificaciones
- OUTPUT2QQDocumento20 páginasOUTPUT2QQLeila Padilla BenitezAún no hay calificaciones
- Blast HitsDocumento7 páginasBlast HitsYaz StrangeAún no hay calificaciones
- Algoritmos GenéticosDocumento30 páginasAlgoritmos GenéticosAlvaro Daniel Castillo Carrera100% (4)
- Resultado Test 3 SuperadoDocumento2 páginasResultado Test 3 Superadoacobo705Aún no hay calificaciones
- Algoritmo GeneticoDocumento44 páginasAlgoritmo GeneticocarlosAún no hay calificaciones
- Cuestionario BioinformáticaDocumento3 páginasCuestionario BioinformáticaDominique HerreraAún no hay calificaciones
- Presentación - KNN.20085205Documento36 páginasPresentación - KNN.20085205Enrique HernandezGarciaAún no hay calificaciones
- Semana 2Documento52 páginasSemana 2Saul Gonzales AlarconAún no hay calificaciones
- Tips - Banco-De-Preg-Psa-Bolivia-Matematica 2013 UAGRMDocumento198 páginasTips - Banco-De-Preg-Psa-Bolivia-Matematica 2013 UAGRMBenjaminAún no hay calificaciones
- Clase 4 Cladismo PDFDocumento68 páginasClase 4 Cladismo PDFAndres AriasAún no hay calificaciones
- Fertilizantes en EsDocumento15 páginasFertilizantes en EsSantiago ToapantaAún no hay calificaciones
- California 2Documento65 páginasCalifornia 2Santiago ToapantaAún no hay calificaciones
- Bioinformatica 4Documento19 páginasBioinformatica 4Santiago ToapantaAún no hay calificaciones
- California 1Documento70 páginasCalifornia 1Santiago ToapantaAún no hay calificaciones
- Bioinformatica 2Documento11 páginasBioinformatica 2Santiago ToapantaAún no hay calificaciones
- Lombriz de Tierra - En.esDocumento8 páginasLombriz de Tierra - En.esSantiago ToapantaAún no hay calificaciones
- Lombriz de Tierra 2.en - EsDocumento5 páginasLombriz de Tierra 2.en - EsSantiago ToapantaAún no hay calificaciones
- Izquierdo 2016 Humedad RiegoDocumento59 páginasIzquierdo 2016 Humedad RiegoSantiago ToapantaAún no hay calificaciones
- Casahualpa Priscila REPORTEDENOCONFORMIDADESDocumento2 páginasCasahualpa Priscila REPORTEDENOCONFORMIDADESSantiago ToapantaAún no hay calificaciones
- Plan de Negocios Salsa de Manzana PicanteDocumento23 páginasPlan de Negocios Salsa de Manzana PicanteSantiago Toapanta100% (1)
- Bioinformatica 5Documento14 páginasBioinformatica 5Santiago ToapantaAún no hay calificaciones
- Control de Lectura Norma Iso 19011. Directrices para La Auditoria. BiotecnologíaDocumento7 páginasControl de Lectura Norma Iso 19011. Directrices para La Auditoria. BiotecnologíaSantiago ToapantaAún no hay calificaciones
- CasahualpaP AplicaciónProgramaProjectDocumento12 páginasCasahualpaP AplicaciónProgramaProjectSantiago ToapantaAún no hay calificaciones
- CasahualpaP - Matriz de Aspectos AmbientalesDocumento8 páginasCasahualpaP - Matriz de Aspectos AmbientalesSantiago ToapantaAún no hay calificaciones
- CasahualpaP LineamientospostercientíficoDocumento4 páginasCasahualpaP LineamientospostercientíficoSantiago ToapantaAún no hay calificaciones
- Análisis Pest ElDocumento2 páginasAnálisis Pest ElSantiago ToapantaAún no hay calificaciones
- CasahualpaP DiseñodeunproyectodeinvestigaciónDocumento3 páginasCasahualpaP DiseñodeunproyectodeinvestigaciónSantiago ToapantaAún no hay calificaciones
- CasahualpaP AplicaciónProgramaProjectDocumento12 páginasCasahualpaP AplicaciónProgramaProjectSantiago ToapantaAún no hay calificaciones
- Cronograma VincuDocumento10 páginasCronograma VincuSantiago ToapantaAún no hay calificaciones
- CasahualpaP DiseñodeunproyectodeinvestigaciónDocumento3 páginasCasahualpaP DiseñodeunproyectodeinvestigaciónSantiago ToapantaAún no hay calificaciones
- CanvasDocumento6 páginasCanvasSantiago ToapantaAún no hay calificaciones
- Cronograma VincuDocumento10 páginasCronograma VincuSantiago ToapantaAún no hay calificaciones
- Plan de Negocios Salsa de Manzana PicanteDocumento23 páginasPlan de Negocios Salsa de Manzana PicanteSantiago Toapanta100% (1)
- CanvasDocumento6 páginasCanvasSantiago ToapantaAún no hay calificaciones
- Encuesta 1Documento2 páginasEncuesta 1Santiago ToapantaAún no hay calificaciones
- CasahualpaP LineamientospostercientíficoDocumento4 páginasCasahualpaP LineamientospostercientíficoSantiago ToapantaAún no hay calificaciones
- CasahualpaP - Matriz de Aspectos AmbientalesDocumento8 páginasCasahualpaP - Matriz de Aspectos AmbientalesSantiago ToapantaAún no hay calificaciones
- Control de Lectura Norma Iso 19011. Directrices para La Auditoria. BiotecnologíaDocumento7 páginasControl de Lectura Norma Iso 19011. Directrices para La Auditoria. BiotecnologíaSantiago ToapantaAún no hay calificaciones
- Análisis Pest ElDocumento2 páginasAnálisis Pest ElSantiago ToapantaAún no hay calificaciones
- AlcalofilosDocumento8 páginasAlcalofilosFernando PinedaAún no hay calificaciones
- Institución Educativa Bachillerato Patía El Bordo Guía # 11 Grado SeptimoDocumento3 páginasInstitución Educativa Bachillerato Patía El Bordo Guía # 11 Grado Septimobrithny lizeth bernal lunaAún no hay calificaciones
- Aplicaciones de Los Animales Genéticamente ModificadosDocumento6 páginasAplicaciones de Los Animales Genéticamente ModificadosLauro Emmanuel Contreras NavarroAún no hay calificaciones
- EVIDENCIA 1 Bioq - Irma FavelaDocumento8 páginasEVIDENCIA 1 Bioq - Irma FavelaIrma FavelaAún no hay calificaciones
- Potencial de Acción Lorena y PaulinaDocumento4 páginasPotencial de Acción Lorena y PaulinaMARISSA PAULINA MOLINA SALAZARAún no hay calificaciones
- Estructura y Función CelularDocumento4 páginasEstructura y Función CelularCarolina OrtizAún no hay calificaciones
- Extraccion y Ensayo de La Actividad Invertasa de Levadura y PanaderiaDocumento1 páginaExtraccion y Ensayo de La Actividad Invertasa de Levadura y PanaderiaAndrea Nikol AlvaradoAún no hay calificaciones
- Clase 13 Marcadores MolecularesDocumento82 páginasClase 13 Marcadores MolecularesDkavid HxAún no hay calificaciones
- Estudio de Mercado - Biodiversidad Paises Andinos PDFDocumento528 páginasEstudio de Mercado - Biodiversidad Paises Andinos PDFferamorosAún no hay calificaciones
- EL DESTINO DEL ESQUELETO CARBONADO (Recuperado Automáticamente)Documento2 páginasEL DESTINO DEL ESQUELETO CARBONADO (Recuperado Automáticamente)Richard JoelAún no hay calificaciones
- Wuolah Free UNIDAD 5Documento20 páginasWuolah Free UNIDAD 5Adrian Dacosta LazeraAún no hay calificaciones
- Biologia GeneralDocumento7 páginasBiologia GeneralNATHALYAún no hay calificaciones
- Q. Orgánica Introduccion A La Quimica HeterociclicaDocumento45 páginasQ. Orgánica Introduccion A La Quimica HeterociclicaAlexis ZavalaAún no hay calificaciones
- METABOLISMO DE LOS NUTRIENTES Resumen BromatologíaDocumento11 páginasMETABOLISMO DE LOS NUTRIENTES Resumen BromatologíaIván Narváez AguilarAún no hay calificaciones
- Ley de BioseguridadDocumento6 páginasLey de BioseguridadJuan Carlos Gonzalez Santillan100% (1)
- Ing Bioquímica TripticoDocumento2 páginasIng Bioquímica TripticoUli RAMOS0% (1)
- Bioenergética de Los Carbohidratos: Digestión y Absorción de Los Carbohidratos. Glicolisis: Incidencia. Vía. Regulación. Balance Bioenergético.Documento23 páginasBioenergética de Los Carbohidratos: Digestión y Absorción de Los Carbohidratos. Glicolisis: Incidencia. Vía. Regulación. Balance Bioenergético.Darwin ValverdeAún no hay calificaciones
- BF Informe N°7Documento12 páginasBF Informe N°7Hikigaya Hachiman100% (1)
- Práctica 1 - Transporte de Glucosa en Levaduras Seccion 2Documento38 páginasPráctica 1 - Transporte de Glucosa en Levaduras Seccion 2Tania Karen Rodriguez GarciaAún no hay calificaciones
- Biologia - Examen - 1ro Bgu - 2do QuimestreDocumento9 páginasBiologia - Examen - 1ro Bgu - 2do QuimestreMateo ReyesAún no hay calificaciones
- Líneas de Investigación UG y Dominio Facultad - UG - 2015-2021Documento1 páginaLíneas de Investigación UG y Dominio Facultad - UG - 2015-2021Julio Zuñiga100% (1)
- Exposición 5 Impacto de La Biotecnología en La AgroindustriaDocumento1 páginaExposición 5 Impacto de La Biotecnología en La AgroindustriaDiego Armando Pérez ReséndizAún no hay calificaciones
- Circuitos de ControlDocumento169 páginasCircuitos de ControlDiana CacuangoAún no hay calificaciones
- ENSAYO Cilclo de Krebs y TranscripcionDocumento2 páginasENSAYO Cilclo de Krebs y TranscripcionAna Victoria Ramírez Iriarte50% (2)
- ElectroforesisDocumento10 páginasElectroforesisAilin GonzalezAún no hay calificaciones
- Clase Hormonas Proteicas - Tipos-Sintesis y Mecanismos de AccionDocumento89 páginasClase Hormonas Proteicas - Tipos-Sintesis y Mecanismos de AccionAlan EscobarAún no hay calificaciones
- Examen B1 Segunda UnidadDocumento4 páginasExamen B1 Segunda UnidadEU GonzalezAún no hay calificaciones
- FolletosDocumento2 páginasFolletossrturoAún no hay calificaciones
- BiologaDocumento2 páginasBiologaSandra Aguirre AngeloffAún no hay calificaciones
- Lípidos (Taller Preguntas de Revisión)Documento3 páginasLípidos (Taller Preguntas de Revisión)Juan Sebastián GirónAún no hay calificaciones