También podría gustarte
- Control de procesos y seguridad e higiene. INAV0109De EverandControl de procesos y seguridad e higiene. INAV0109Aún no hay calificaciones
- MF0297_2 - Elaboración de preparados cárnicos frescosDe EverandMF0297_2 - Elaboración de preparados cárnicos frescosCalificación: 5 de 5 estrellas5/5 (1)
- 8218 Detergente Terminado Roma FTDocumento2 páginas8218 Detergente Terminado Roma FTMoshe MontfordAún no hay calificaciones
- Contrato Prestacion de ServiciosDocumento3 páginasContrato Prestacion de ServiciosVanessa Barrios100% (1)
- Acta ConstitutivaDocumento10 páginasActa ConstitutivaYudi PequeAún no hay calificaciones
- GUÍA PARA LA GESTIÓN DE CALIDAD Y LA SEGURIDAD LABORAL DE LOS SERVICIOS DE SALUD: Versión global aplicable a todo sistema de saludDe EverandGUÍA PARA LA GESTIÓN DE CALIDAD Y LA SEGURIDAD LABORAL DE LOS SERVICIOS DE SALUD: Versión global aplicable a todo sistema de saludCalificación: 5 de 5 estrellas5/5 (2)
- Comisión del Codex Alimentarius: Manual de Procedimiento 26 edicionDe EverandComisión del Codex Alimentarius: Manual de Procedimiento 26 edicionAún no hay calificaciones
- Instrumento de evaluación del sistema de control de los alimentos: Introducción y glosarioDe EverandInstrumento de evaluación del sistema de control de los alimentos: Introducción y glosarioAún no hay calificaciones
- 8218 Det. Terminado Bca Nieves HDSDocumento4 páginas8218 Det. Terminado Bca Nieves HDSMoshe Montford100% (5)
- 8218 Det. Terminado Bca Nieves HDSDocumento4 páginas8218 Det. Terminado Bca Nieves HDSMoshe Montford100% (5)
- HSPT-6050505 Cloralex El Rendidor GelDocumento8 páginasHSPT-6050505 Cloralex El Rendidor GelMoshe MontfordAún no hay calificaciones
- 8218 Pastilla Zote Blanco 400G FTDocumento2 páginas8218 Pastilla Zote Blanco 400G FTMoshe Montford100% (1)
- Nom 164 Ssa1 1998Documento12 páginasNom 164 Ssa1 1998nagato1119Aún no hay calificaciones
- 8218 Aceite 1 2 3 HDSDocumento5 páginas8218 Aceite 1 2 3 HDSMoshe Montford100% (1)
- El Contrato PsicológicoDocumento12 páginasEl Contrato Psicológicojuan carlos espinal uribeAún no hay calificaciones
- Nom 241 Ssa1 2021 1635274617Documento104 páginasNom 241 Ssa1 2021 1635274617Jesús RoblesAún no hay calificaciones
- Instrumento de evaluación del sistema de control de los alimentos: Dimensión D - Base científica/de conocimientos y mejoramiento continuoDe EverandInstrumento de evaluación del sistema de control de los alimentos: Dimensión D - Base científica/de conocimientos y mejoramiento continuoAún no hay calificaciones
- Higiene general en la industria alimentaria. INAQ0108De EverandHigiene general en la industria alimentaria. INAQ0108Aún no hay calificaciones
- NOM-164-SSA1-2015, Buenas Prácticas de Fabricación de FármacosDocumento49 páginasNOM-164-SSA1-2015, Buenas Prácticas de Fabricación de Fármacosstenotrophomonas maltophiliaAún no hay calificaciones
- NOM-164-SSA1-2015, Buenas Prácticas de Fabricación de Fármacos.Documento41 páginasNOM-164-SSA1-2015, Buenas Prácticas de Fabricación de Fármacos.Adinha TamayoAún no hay calificaciones
- Derecho Comercial de MedinaDocumento14 páginasDerecho Comercial de MedinaAtaraxia Dark67% (3)
- Resumen ContratosDocumento33 páginasResumen ContratosAna100% (3)
- Nom 164Documento49 páginasNom 164laura esquivelAún no hay calificaciones
- NOM-164-SSA1-2015 BPF FármDocumento40 páginasNOM-164-SSA1-2015 BPF FármMauro Alfredo Calzadilla GonzálezAún no hay calificaciones
- NORMA Oficial Mexicana NOM-164-SSA1-2015, Buenas Prácticas de Fabricación de Fármacos.Documento44 páginasNORMA Oficial Mexicana NOM-164-SSA1-2015, Buenas Prácticas de Fabricación de Fármacos.Adrian GedioneAún no hay calificaciones
- DOF - Diario Oficial de La FederaciónDocumento46 páginasDOF - Diario Oficial de La FederaciónAlfredo ArroyoAún no hay calificaciones
- Nom 059 Ssa1 2015Documento69 páginasNom 059 Ssa1 2015Girard GarciaAún no hay calificaciones
- NOM 164 SSA1 2015 - BPF - FarmacosDocumento41 páginasNOM 164 SSA1 2015 - BPF - FarmacosaarroyosAún no hay calificaciones
- NORMA Oficial Mexicana NOM-249-SSA1-2010, Mezclas Estériles - Nutricionales y Medicamentosas, e Instalaciones para Su PreparaciónDocumento23 páginasNORMA Oficial Mexicana NOM-249-SSA1-2010, Mezclas Estériles - Nutricionales y Medicamentosas, e Instalaciones para Su PreparaciónFrancisco Alonso VázquezAún no hay calificaciones
- NOM-249-SSA1-2010, Mezclas EstérilesDocumento19 páginasNOM-249-SSA1-2010, Mezclas EstérilesZhadow TorchonAún no hay calificaciones
- DOF - Diario Oficial de La FederaciónDocumento59 páginasDOF - Diario Oficial de La FederaciónCARLOS ALBERTO VITE SIERRAAún no hay calificaciones
- NOM 241 SSA1 2012 - 23abr2012Documento46 páginasNOM 241 SSA1 2012 - 23abr2012lindajosefinaAún no hay calificaciones
- Nom 164 Ssa1 2015Documento38 páginasNom 164 Ssa1 2015Juan Ramon Castro SanchezAún no hay calificaciones
- Nom 241 Ssa1 2021 BPM Pará Dispositivos MédicosDocumento59 páginasNom 241 Ssa1 2021 BPM Pará Dispositivos MédicosCarlos CarrilloAún no hay calificaciones
- Norma 164Documento42 páginasNorma 164Briz ArroyoAún no hay calificaciones
- Nom 164 Ssa1 2015Documento38 páginasNom 164 Ssa1 2015Gabriela Cecilia Castro CastroAún no hay calificaciones
- Nom 241 Ssa1 2012Documento30 páginasNom 241 Ssa1 2012validadorAún no hay calificaciones
- DOF - Diario Oficial de La FederaciónDocumento80 páginasDOF - Diario Oficial de La FederaciónJess RamosAún no hay calificaciones
- NOM 059 2015 PresentaciónDocumento86 páginasNOM 059 2015 PresentaciónHaBa TorNaAún no hay calificaciones
- Anteproyecto PROY NOM 241 SSA1 2017 Versión FirmadaDocumento64 páginasAnteproyecto PROY NOM 241 SSA1 2017 Versión FirmadaFede FerAún no hay calificaciones
- NORMA Oficial Mexicana NOM-241-SSA1-2012Documento103 páginasNORMA Oficial Mexicana NOM-241-SSA1-2012MIGUEL ANGEL CASTRO LEALAún no hay calificaciones
- Norma 059Documento86 páginasNorma 059Briz ArroyoAún no hay calificaciones
- Nom 059Documento112 páginasNom 059kronostonhypnosAún no hay calificaciones
- Monografias de Bolsas para Esterilizar FeumDocumento54 páginasMonografias de Bolsas para Esterilizar FeumJuan TorresAún no hay calificaciones
- Buenas Prácticas de Fabricación de Medicamentos.Documento69 páginasBuenas Prácticas de Fabricación de Medicamentos.Pash CruzAún no hay calificaciones
- GMP para Dispositivos MédicosDocumento72 páginasGMP para Dispositivos MédicosGermán Cárdenas AlvarezAún no hay calificaciones
- Qué Es La NOM 059Documento4 páginasQué Es La NOM 059Rafael Lopez PAún no hay calificaciones
- NOM-241-SSA1-2012 - Buenas Prácticas de Fabricación para Establecimientos Dedicados A La Fabricación de Dispositivos MédicosDocumento28 páginasNOM-241-SSA1-2012 - Buenas Prácticas de Fabricación para Establecimientos Dedicados A La Fabricación de Dispositivos MédicoslmsabogaloAún no hay calificaciones
- NORMA 220 FarmacovigilanciaDocumento18 páginasNORMA 220 FarmacovigilanciaCatherine GerardAún no hay calificaciones
- Dispo 3602-18 NuevaGMP ANMATDocumento246 páginasDispo 3602-18 NuevaGMP ANMATFernando Sebastian HussAún no hay calificaciones
- Nom-001-Ssa1-2010. FeumDocumento6 páginasNom-001-Ssa1-2010. FeumLore ORtiizzAún no hay calificaciones
- NOM-241-SSA1-2012, Buenas Prácticas de Fabricación Dispositivos MédicosDOFDocumento30 páginasNOM-241-SSA1-2012, Buenas Prácticas de Fabricación Dispositivos MédicosDOFfuentenaturaAún no hay calificaciones
- 164 Ssa 1Documento41 páginas164 Ssa 1DrayDergonAún no hay calificaciones
- Nom 241Documento34 páginasNom 241Manuel TorresAún no hay calificaciones
- NORMA Oficial Mexicana NOM-001-SSA1-2020Documento9 páginasNORMA Oficial Mexicana NOM-001-SSA1-2020Jose G VegaAún no hay calificaciones
- Nom 220 Ssa1 2016 PDFDocumento21 páginasNom 220 Ssa1 2016 PDFAle QuirozAún no hay calificaciones
- Nom 220 Ssa1 2016Documento24 páginasNom 220 Ssa1 2016Gabriel Aldape ArtegaAún no hay calificaciones
- Nom 159 Ssa1 2016 Nom 159 Dof - 20180719 20180116Documento14 páginasNom 159 Ssa1 2016 Nom 159 Dof - 20180719 20180116Edrian Fernando Rebollo GutiérrezAún no hay calificaciones
- Nom 220 Ssa1 2016Documento19 páginasNom 220 Ssa1 2016Girard GarciaAún no hay calificaciones
- Nom 241 Ssa1 2012Documento38 páginasNom 241 Ssa1 2012FatimaAún no hay calificaciones
- Nom 240 Ssa1 2012Documento15 páginasNom 240 Ssa1 2012susanaAún no hay calificaciones
- Norma 073Documento24 páginasNorma 073Arturo BelmontAún no hay calificaciones
- NOM159SSAI2016 NOMs Huevo SALUD - 20190121 20180116Documento10 páginasNOM159SSAI2016 NOMs Huevo SALUD - 20190121 20180116Nancy OlivaresAún no hay calificaciones
- Norma Oficial Mexicana Nom 164 Ssa1 2015 Buenas Prcticas de Fabricacin de FrmacosDocumento77 páginasNorma Oficial Mexicana Nom 164 Ssa1 2015 Buenas Prcticas de Fabricacin de FrmacosManuel Alejandro Iza GutierrezAún no hay calificaciones
- Nom 176-1998Documento9 páginasNom 176-1998Adriana Cinthya LRAún no hay calificaciones
- HuevoDocumento10 páginasHuevoAbdiel PucAún no hay calificaciones
- Instrumento de evaluación del sistema de control de los alimentos: Dimensión A - Aportaciones y recursosDe EverandInstrumento de evaluación del sistema de control de los alimentos: Dimensión A - Aportaciones y recursosAún no hay calificaciones
- Instrumento de evaluación del sistema de control de los alimentos: Dimensión B - Funciones de controlDe EverandInstrumento de evaluación del sistema de control de los alimentos: Dimensión B - Funciones de controlAún no hay calificaciones
- Seguridad e Higiene en un obrador de panadería y bollería. INAF0108De EverandSeguridad e Higiene en un obrador de panadería y bollería. INAF0108Aún no hay calificaciones
- Ciencia regulatoria: Medicamentos bio y su relevancia para la saludDe EverandCiencia regulatoria: Medicamentos bio y su relevancia para la saludAún no hay calificaciones
- Aplicación de normas y condiciones higiénico-sanitarias en restauración. HOTR0108De EverandAplicación de normas y condiciones higiénico-sanitarias en restauración. HOTR0108Aún no hay calificaciones
- Listado de Insumos para La Salud Considerados Como de Bajo Riesgo para Efectos de Obtención Del Registro Sanitario.Documento49 páginasListado de Insumos para La Salud Considerados Como de Bajo Riesgo para Efectos de Obtención Del Registro Sanitario.Moshe MontfordAún no hay calificaciones
- Plantilla de Evaluacion Del EmpleadoDocumento7 páginasPlantilla de Evaluacion Del EmpleadoKaren TrujilloAún no hay calificaciones
- Reg Lgs Mcsaeps 281204Documento186 páginasReg Lgs Mcsaeps 281204Moshe MontfordAún no hay calificaciones
- FORMATOSEVA2011Documento13 páginasFORMATOSEVA2011Luis Felipe Martínez GarcíaAún no hay calificaciones
- REGLAMENTO de Insumos para La Salud.Documento41 páginasREGLAMENTO de Insumos para La Salud.Moshe MontfordAún no hay calificaciones
- Pendiestes de Mentanimiento Julio 2022Documento4 páginasPendiestes de Mentanimiento Julio 2022Moshe MontfordAún no hay calificaciones
- SDS IDH 2569954 - 123 Multiusos Det PoDocumento11 páginasSDS IDH 2569954 - 123 Multiusos Det PoMoshe MontfordAún no hay calificaciones
- "Tierra Y Libertad": Periódico OficialDocumento12 páginas"Tierra Y Libertad": Periódico OficialmrscribdAún no hay calificaciones
- HSPT Ácido Muriático Sultán Aroma LimónDocumento7 páginasHSPT Ácido Muriático Sultán Aroma LimónMoshe MontfordAún no hay calificaciones
- 8218 Aceite 1 2 3 FTDocumento1 página8218 Aceite 1 2 3 FTMoshe MontfordAún no hay calificaciones
- HSPT Ácido Muriático Sultán Aroma LimónDocumento7 páginasHSPT Ácido Muriático Sultán Aroma LimónMoshe MontfordAún no hay calificaciones
- 8218 Pastilla Zote Blanco 400G HDSDocumento4 páginas8218 Pastilla Zote Blanco 400G HDSMoshe MontfordAún no hay calificaciones
- 8218 Pastilla Rosa Venus Blanco 100g HdsDocumento4 páginas8218 Pastilla Rosa Venus Blanco 100g HdsMoshe MontfordAún no hay calificaciones
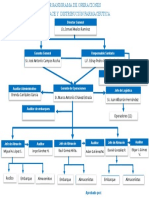
- Organigrama OperacionesDocumento1 páginaOrganigrama OperacionesMoshe MontfordAún no hay calificaciones
- 8218 Pastilla Rosa Venus Blanco 100g FTDocumento2 páginas8218 Pastilla Rosa Venus Blanco 100g FTMoshe Montford100% (1)
- La Forma y Los FormalismosDocumento5 páginasLa Forma y Los FormalismosKENIA JAQUELIN DIMAS MARROQUINAún no hay calificaciones
- Contrato de Trabajo PlanillaDocumento3 páginasContrato de Trabajo PlanillaJose MoralesAún no hay calificaciones
- El Derecho ChinoDocumento14 páginasEl Derecho ChinojeffryAún no hay calificaciones
- Contrato ArrendamientiDocumento4 páginasContrato Arrendamientiaranxa robledo montelongoAún no hay calificaciones
- Contrato de Prestamo de Dinero Pajuelo Macedomayo19Documento2 páginasContrato de Prestamo de Dinero Pajuelo Macedomayo19Dario RamosAún no hay calificaciones
- Modelo de ContratoDocumento3 páginasModelo de ContratoMilciadesAún no hay calificaciones
- Contrato de Trabajo A Término FijoDocumento7 páginasContrato de Trabajo A Término FijoNayvis Navarro FonsecaAún no hay calificaciones
- Addendum Contrato de Compraventa DefinitivaDocumento4 páginasAddendum Contrato de Compraventa DefinitivaLicda Milena PoloAún no hay calificaciones
- La AnticresisDocumento6 páginasLa AnticresisLuis Rodriguez AsorzaAún no hay calificaciones
- Temas 1.1-1.4 (Unidad 1)Documento18 páginasTemas 1.1-1.4 (Unidad 1)fernanda sanzAún no hay calificaciones
- 3 Limites A La Aplicacion Del Principio Del La Primacía de La Realidad. Lora PDFDocumento13 páginas3 Limites A La Aplicacion Del Principio Del La Primacía de La Realidad. Lora PDFjuliaAún no hay calificaciones
- Contrato de Prestamo 1Documento3 páginasContrato de Prestamo 1Daniel CalderonAún no hay calificaciones
- Capitulo I Generalidades Del Derecho Laboral (Actualizado 2018)Documento28 páginasCapitulo I Generalidades Del Derecho Laboral (Actualizado 2018)carolina arias100% (1)
- Contrato de Seguro EscritoDocumento13 páginasContrato de Seguro EscritoAna Karina Díaz MercadoAún no hay calificaciones
- 5 Fallo Rojas Con JunjiDocumento32 páginas5 Fallo Rojas Con JunjimiguelvasoAún no hay calificaciones
- Clase 02 - Ley 29946 - 20171220131008Documento36 páginasClase 02 - Ley 29946 - 20171220131008Martha DiazAún no hay calificaciones
- CONTRATO COLECTIVO EQUIPO 2 (1) (1) (Autoguardado)Documento10 páginasCONTRATO COLECTIVO EQUIPO 2 (1) (1) (Autoguardado)Alexandra BledelAún no hay calificaciones
- Image 20240326 0001Documento5 páginasImage 20240326 0001georgenicosoto1Aún no hay calificaciones
- Importancia Del Principio de Congruencia Procesal en La Actividad Judicial y El Papel Del Ministerio PúblicoDocumento6 páginasImportancia Del Principio de Congruencia Procesal en La Actividad Judicial y El Papel Del Ministerio PúblicoalbertochinoAún no hay calificaciones
- Terminacion de Contratos de TrabajoDocumento18 páginasTerminacion de Contratos de TrabajoCARMENHP1978Aún no hay calificaciones
- CIVIL Juzgado Tercero Civil PROCEDENTE 31 2021Documento15 páginasCIVIL Juzgado Tercero Civil PROCEDENTE 31 2021JOSE BENITEZAún no hay calificaciones
- Cuaderno de Obra 04.okDocumento18 páginasCuaderno de Obra 04.okGaby Escobar FalconAún no hay calificaciones
- Ejem Contrato Civil ObraDocumento3 páginasEjem Contrato Civil Obramilena bermudezAún no hay calificaciones
- Introducción A La Contabilidad GanaderaDocumento20 páginasIntroducción A La Contabilidad GanaderaAlejandra GomezAún no hay calificaciones
- Taller CiudadaníaDocumento78 páginasTaller CiudadaníaElisa BaglioniAún no hay calificaciones