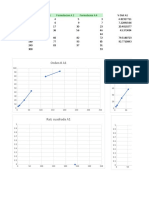
También podría gustarte
- Listado 2Documento83 páginasListado 2Auxiliar SistemasAún no hay calificaciones
- Reporte InventarioDocumento98 páginasReporte InventarioDaniel AltamiranoAún no hay calificaciones
- Pno-Far-01 Elaboracion de Pno V.1.Documento8 páginasPno-Far-01 Elaboracion de Pno V.1.Daniel Altamirano100% (1)
- PNO MuestraDocumento3 páginasPNO MuestraDaniel AltamiranoAún no hay calificaciones
- Tabla de Diluciones.1Documento9 páginasTabla de Diluciones.1Daniel AltamiranoAún no hay calificaciones
- Seguimiento de La Unidad de Farmacovigilancia (Agrupado)Documento12 páginasSeguimiento de La Unidad de Farmacovigilancia (Agrupado)Daniel AltamiranoAún no hay calificaciones
- Compendio de ActividadesDocumento4 páginasCompendio de ActividadesDaniel AltamiranoAún no hay calificaciones
- XDXDXDDocumento3 páginasXDXDXDDaniel AltamiranoAún no hay calificaciones
- Morfología y Fisiología de Los EritrocitosDocumento2 páginasMorfología y Fisiología de Los EritrocitosDaniel AltamiranoAún no hay calificaciones
- CUESTIONARIODocumento4 páginasCUESTIONARIOAnna María RivasAún no hay calificaciones
- Ejercicios de ConversionDocumento18 páginasEjercicios de ConversionROMERO BARBOZA KIARA ESTHEFANYAún no hay calificaciones
- DispensaciónDocumento10 páginasDispensaciónFABIO ALBERTO BRIJALDO RAMIREZAún no hay calificaciones
- IVERMECTINADocumento6 páginasIVERMECTINAAnys Canela0% (1)
- Farmacognosia 2013-2014Documento23 páginasFarmacognosia 2013-2014Juan Carlos Ríos MartínezAún no hay calificaciones
- Presentación de EmbriaguezDocumento8 páginasPresentación de EmbriaguezMary Luz DuqueAún no hay calificaciones
- Medicamentos AntipsicoticosDocumento8 páginasMedicamentos AntipsicoticosNeliza Borda huamanAún no hay calificaciones
- Laxantes en Perros y Gatos-Nathaly Sinchi-5to BDocumento8 páginasLaxantes en Perros y Gatos-Nathaly Sinchi-5to BLalitaSinchiAún no hay calificaciones
- Todas Las Resolucion Del PosDocumento1117 páginasTodas Las Resolucion Del PosluisAún no hay calificaciones
- Formulario Nacional 2022Documento573 páginasFormulario Nacional 2022Raquel Gonzalez Rodriguez100% (1)
- Dolor PostoperatorioDocumento44 páginasDolor Postoperatorioginna vargasAún no hay calificaciones
- Lemke-Farmacos Hidrofilicos-FinalDocumento6 páginasLemke-Farmacos Hidrofilicos-FinalRios MercedesAún no hay calificaciones
- Pedido Recibido de Droaliado El 01 de Octubre Del 2020Documento7 páginasPedido Recibido de Droaliado El 01 de Octubre Del 2020angie michelle castroAún no hay calificaciones
- FARMACOS BDocumento3 páginasFARMACOS BMónica Alejandra Estrada SorianoAún no hay calificaciones
- 2do Parcial Fármacologia - FlorezDocumento74 páginas2do Parcial Fármacologia - FlorezLunita FredesAún no hay calificaciones
- Cartilla FarmacologíaDocumento137 páginasCartilla FarmacologíaEloisa FreyreAún no hay calificaciones
- Derivados Del Ácido Antranílico.Documento3 páginasDerivados Del Ácido Antranílico.Stalyn JoséAún no hay calificaciones
- Semana 16 Farmacias InclusivasDocumento31 páginasSemana 16 Farmacias InclusivasLuis Taipe SanchezAún no hay calificaciones
- MANZANILLADocumento10 páginasMANZANILLADayana BecerraAún no hay calificaciones
- Bioequivalentes Mayo 2023Documento6 páginasBioequivalentes Mayo 2023Claudia ChávezAún no hay calificaciones
- 53505-Texto Del Artículo-164618-1-10-20210610Documento16 páginas53505-Texto Del Artículo-164618-1-10-20210610Yleyam RamirezAún no hay calificaciones
- Capítulo 4. Toxicidad de Fármacos y EnvenenamientoDocumento6 páginasCapítulo 4. Toxicidad de Fármacos y EnvenenamientoGABRIEL MOCTEZUMAAún no hay calificaciones
- BETA MICOTER Crema 10 - 0,5 MG - Datos GeneralesDocumento11 páginasBETA MICOTER Crema 10 - 0,5 MG - Datos GeneralesheraclitodeefesoAún no hay calificaciones
- INFORME TÉCNICO LoperamidaDocumento4 páginasINFORME TÉCNICO LoperamidaMiguel Oviedo SanchezAún no hay calificaciones
- Ficha de Solicitud de Antibiograma FormularioDocumento2 páginasFicha de Solicitud de Antibiograma FormularioFreddy SerrudoAún no hay calificaciones
- Bianca Notas FarmaDocumento177 páginasBianca Notas FarmaGerardo SanchezAún no hay calificaciones
- Seminario AntihipertensivosDocumento7 páginasSeminario AntihipertensivoswallpapersAún no hay calificaciones
- Control de Calidad de Comprimidos GenéricosDocumento43 páginasControl de Calidad de Comprimidos GenéricosCHRISTIAN ALEJANDRO LEAL ALVAREZAún no hay calificaciones
- Salud y Fármacos ArtículoDocumento76 páginasSalud y Fármacos ArtículojavierocuAún no hay calificaciones