También podría gustarte
- Usermanual ARC EASEFocus EspagnolDocumento28 páginasUsermanual ARC EASEFocus EspagnolFranco CarrilloAún no hay calificaciones
- 2ESOLibroCompleto PDFDocumento153 páginas2ESOLibroCompleto PDFMar RevueltoAún no hay calificaciones
- P04 CastellanoDocumento6 páginasP04 CastellanoFrak pepinoAún no hay calificaciones
- El Paradigma de La Complejidad Como Salida de La Crisis de La PosmodernidadDocumento14 páginasEl Paradigma de La Complejidad Como Salida de La Crisis de La PosmodernidadescherichioAún no hay calificaciones
- Clases de KefirDocumento7 páginasClases de KefirMarta Papaseit LimónAún no hay calificaciones
- TP3 ProblemasVMNADocumento3 páginasTP3 ProblemasVMNAescherichioAún no hay calificaciones
- Volumetria en Medio No AcuosoDocumento16 páginasVolumetria en Medio No Acuosoescherichio100% (1)
- 4 VinilbenzoicoDocumento4 páginas4 VinilbenzoicoescherichioAún no hay calificaciones
- Acido PercloricoDocumento2 páginasAcido PercloricoescherichioAún no hay calificaciones
- ValoracionesDocumento6 páginasValoracionesescherichioAún no hay calificaciones
- Fuerza IonicaDocumento2 páginasFuerza IonicaescherichioAún no hay calificaciones
- (Taller - Windows) 1 - 2Documento5 páginas(Taller - Windows) 1 - 2juan sebastianAún no hay calificaciones
- Trucos W10Documento63 páginasTrucos W10annyAún no hay calificaciones
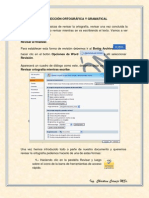
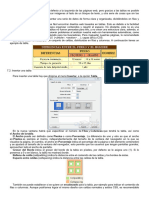
- Corrección Ortográfica y GramaticalDocumento4 páginasCorrección Ortográfica y GramaticalchriscornejoAún no hay calificaciones
- Anexo Manual PowerPointDocumento9 páginasAnexo Manual PowerPointpepe008327Aún no hay calificaciones
- Atajos de Teclado en ExcelDocumento8 páginasAtajos de Teclado en ExcelEstrada HarryAún no hay calificaciones
- Ceballos: Java 2 - Lenguaje y AplicacionesDocumento30 páginasCeballos: Java 2 - Lenguaje y AplicacionesFco. Javier Ceballos Sierra87% (23)
- Manual Word 2016Documento76 páginasManual Word 2016PAOLA CAROLINA GONZALEZ RUIZAún no hay calificaciones
- Métodos Abreviados en ExcelDocumento3 páginasMétodos Abreviados en ExcelAngie RamosAún no hay calificaciones
- Herramientas de Edición de WORDDocumento3 páginasHerramientas de Edición de WORDThelma Andalon67% (6)
- Test de Conocimientos Sobre OfimáticaDocumento6 páginasTest de Conocimientos Sobre OfimáticaSalmaAún no hay calificaciones
- Guía Técnica - ADMDocumento29 páginasGuía Técnica - ADMGustavo FernandezAún no hay calificaciones
- Tablas DreamweaverDocumento2 páginasTablas DreamweaverCristopher Moquillaza LevanoAún no hay calificaciones
- (Manual) CYPE - Fontanería, Gas, Incendio, Saneamiento, Teleco, Electricidad, Clima (66 Págs) PDFDocumento66 páginas(Manual) CYPE - Fontanería, Gas, Incendio, Saneamiento, Teleco, Electricidad, Clima (66 Págs) PDFNunoAún no hay calificaciones
- Documento de Apoyo para Web2board - v1Documento2 páginasDocumento de Apoyo para Web2board - v1Javier Alex DiazAún no hay calificaciones
- Introducción A Dragonfish Color Y TalleDocumento224 páginasIntroducción A Dragonfish Color Y TalleLyel Augusto0% (1)
- Antologia Herramientas Computacionales IIver2Documento138 páginasAntologia Herramientas Computacionales IIver2amelia berenice estrada100% (1)
- 3.1 1.3.a.configuracion Variable Path PDFDocumento3 páginas3.1 1.3.a.configuracion Variable Path PDFWilliam Alexander JosueAún no hay calificaciones
- Bajar Mega Con IDMDocumento7 páginasBajar Mega Con IDMstgomAún no hay calificaciones
- Informe BingoDocumento32 páginasInforme BingoVALENTINA CABRERA MUÑOZAún no hay calificaciones
- Manual de Referencia MDT v4Documento758 páginasManual de Referencia MDT v4PepeCAD100% (3)
- Word Nivell MigDocumento35 páginasWord Nivell MigFrancisco Javier Hernandez CAún no hay calificaciones
- 04 Manual Basico de PublisherDocumento62 páginas04 Manual Basico de PublisherSucely HernandezAún no hay calificaciones
- Tutorial para Usar RadiobossDocumento8 páginasTutorial para Usar RadiobossjoseAún no hay calificaciones
- 5 Biorresonancia OracionesDocumento22 páginas5 Biorresonancia OracionesNachoRiveroAún no hay calificaciones
- Instalacion y Configuracion de Client Access ExpressDocumento15 páginasInstalacion y Configuracion de Client Access ExpresscesaryngwieAún no hay calificaciones
- Cierre Definitivo SiigoDocumento7 páginasCierre Definitivo Siigoandres ramosAún no hay calificaciones
- Manual CEDAPDocumento6 páginasManual CEDAPMichael Alexander Ortiz CampiñoAún no hay calificaciones
- Dreamweaver8 Portable - Ejercicio de Aplicación BásicaDocumento65 páginasDreamweaver8 Portable - Ejercicio de Aplicación BásicaJaime EcheverriAún no hay calificaciones
- Manual FactuSolDocumento320 páginasManual FactuSoldanielspel100% (1)