También podría gustarte
- 199 Caso Clinico Mordida AbiertaDocumento9 páginas199 Caso Clinico Mordida Abiertamoralk05Aún no hay calificaciones
- Intervencion Logopedica Retraso Evolutivo LenguajeDocumento16 páginasIntervencion Logopedica Retraso Evolutivo Lenguajemoralk05100% (1)
- Espinoza Samaniego Soto Componente LenguajeDocumento110 páginasEspinoza Samaniego Soto Componente Lenguajemoralk05Aún no hay calificaciones
- Curriculum Vitae Tria DoDocumento49 páginasCurriculum Vitae Tria Domoralk05Aún no hay calificaciones
- 17 04 SilvaDocumento11 páginas17 04 Silvamoralk05Aún no hay calificaciones
- Mejorando La Formacion y El Desarrollo Profesional Docente en Latinoamerica. Vaillant D-1Documento16 páginasMejorando La Formacion y El Desarrollo Profesional Docente en Latinoamerica. Vaillant D-1moralk05Aún no hay calificaciones
- El Lenguaje en Educ Infantil-Cep EldaDocumento31 páginasEl Lenguaje en Educ Infantil-Cep Eldamoralk05Aún no hay calificaciones
- FITOREVELADORESDocumento5 páginasFITOREVELADORESLuis Carlos Camargo RodriguezAún no hay calificaciones
- Consecuencias Del Deterioro AmbientalDocumento20 páginasConsecuencias Del Deterioro AmbientalJORGE RIVASAún no hay calificaciones
- Poeg PrimeroDocumento45 páginasPoeg PrimeroyuliethperAún no hay calificaciones
- Métodos de Separación y Extracción de AnalitosDocumento5 páginasMétodos de Separación y Extracción de AnalitosAthenas A.PAún no hay calificaciones
- Ensayo Primer Borrador.Documento6 páginasEnsayo Primer Borrador.lornapazAún no hay calificaciones
- Poliza Completa 253604Documento3 páginasPoliza Completa 253604Santiago SchatzelAún no hay calificaciones
- NUTRICION Clase 4 TerminadoDocumento7 páginasNUTRICION Clase 4 TerminadoANMI Al FinAún no hay calificaciones
- Ruedas de La Vida AutocoachingDocumento29 páginasRuedas de La Vida AutocoachingHernanAún no hay calificaciones
- Taller de Lectura CriticaDocumento5 páginasTaller de Lectura CriticaLeidy SalebeAún no hay calificaciones
- Teoria 13Documento29 páginasTeoria 13Maria AquinoAún no hay calificaciones
- Amparo Indirecto Actualizado BuenoDocumento5 páginasAmparo Indirecto Actualizado BuenoMonica100% (1)
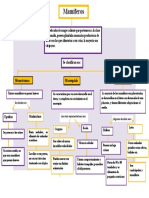
- Mapa Conceptual Mamiferos.1Documento1 páginaMapa Conceptual Mamiferos.1Ale Camargo67% (9)
- Primera Entrega Proyecto Comportamiento OrganizacionalDocumento9 páginasPrimera Entrega Proyecto Comportamiento OrganizacionalMariselaAún no hay calificaciones
- Factores Físicos y Psíquicos, y Componentes Del Sistema de La Memoria.Documento1 páginaFactores Físicos y Psíquicos, y Componentes Del Sistema de La Memoria.Adriana RodriguezAún no hay calificaciones
- Calderon Apaza Michael AlexDocumento10 páginasCalderon Apaza Michael AlexAlex Michael CAAún no hay calificaciones
- Teratología, Agentes FetotóxicosDocumento16 páginasTeratología, Agentes FetotóxicosJosé Luis Majano MurilloAún no hay calificaciones
- PapelDocumento13 páginasPapelNere BattauzAún no hay calificaciones
- Nota Urgencias 33Documento1 páginaNota Urgencias 33Luis NoriegaAún no hay calificaciones
- Rio HuauraDocumento1 páginaRio HuauraBrendi VilleAún no hay calificaciones
- Como Realizar Un Examen Proctologico.Documento2 páginasComo Realizar Un Examen Proctologico.sheylliciousp100% (1)
- Pr-Tal-006 Procedimiento de Fabricacion de Spools de Tuberias AereasDocumento21 páginasPr-Tal-006 Procedimiento de Fabricacion de Spools de Tuberias AereasFrancisco Imaz100% (1)
- Resumen InmunologiaDocumento3 páginasResumen InmunologiaDaniel SalinasAún no hay calificaciones
- Presentación de ColostomiaDocumento34 páginasPresentación de ColostomiaToñaLeón100% (1)
- Microbiología - Wikipedia, La Enciclopedia LibreDocumento7 páginasMicrobiología - Wikipedia, La Enciclopedia LibreReyna MalpasoAún no hay calificaciones
- Cuadro Médico Adeslas MUFACE PontevedraDocumento132 páginasCuadro Médico Adeslas MUFACE PontevedraJose PradoAún no hay calificaciones
- Sistematica de InsectosDocumento116 páginasSistematica de InsectosJuan Carlos Ojeda Cardenas100% (1)
- Practica Final de Psicologia Clinica 2 FHHDJSHDJJJJDocumento12 páginasPractica Final de Psicologia Clinica 2 FHHDJSHDJJJJEmily GracianoAún no hay calificaciones
- CL040-1274-PTL-CI-ISOSIE-005-1015 - 0 Protocolo Hormigonado Losa Ductos GILDocumento1 páginaCL040-1274-PTL-CI-ISOSIE-005-1015 - 0 Protocolo Hormigonado Losa Ductos GILLuis Marín DíazAún no hay calificaciones
- Clase 5. Contaminación Por Partículas en SuspensiónDocumento13 páginasClase 5. Contaminación Por Partículas en SuspensiónDaniel VargasAún no hay calificaciones
- Veterinaria: GradoDocumento4 páginasVeterinaria: GradoAlejandra VisbalAún no hay calificaciones