Documentos de Académico
Documentos de Profesional
Documentos de Cultura
Tema II PDF
Cargado por
Violeta Matdamas MendezTítulo original
Derechos de autor
Formatos disponibles
Compartir este documento
Compartir o incrustar documentos
¿Le pareció útil este documento?
¿Este contenido es inapropiado?
Denunciar este documentoCopyright:
Formatos disponibles
Tema II PDF
Cargado por
Violeta Matdamas MendezCopyright:
Formatos disponibles
GUA DE ESTUDIO
MODALIDAD ADI
TEMA No 2.
SISTEMA DE COMPOSICIN VARIABLE.
COMPORTAMIENTO IDEAL.
Profesora:
Ing. Koralys Goita
2
SISTEMA DE COMPOSICIN VARIABLE
COMPORTAMIENTO IDEAL.
Los sistemas de composicin variable, son conocidos comnmente como
mezclas de dos o ms componentes. En la naturaleza, la materia prima que se
procesa para obtener productos finales se encuentra en su mayora como
mezclas, las cuales deben ser procesadas para obtener con mayor grado de
pureza el producto que se desee. De all, la gran importancia del estudio de los
sistemas de composicin variable, sin sus propiedades seria una tarea difcil
disear equipos, para procesar dicha materia prima.
En esta unidad temtica se estudiar cmo determinarle las propiedades
termodinmicas a este tipo de sistemas, considerando el comportamiento ms
simple (comportamiento ideal). El comportamiento ideal, representa en muchas
ocasiones mezclas de compuestos en un proceso real, presentando interacciones
atmicas o moleculares entre los componentes de la mezcla en los cuales las
fuerzas intermoleculares son casi iguales y simtricas. En este tema, se
estudiaran la mezcla de gases ideales, representando por la ley de Raolt,
obtenindose de este modo un estado de referencia para las mezclas reales
(tema 3). Del mismo modo ocurrir con las soluciones ideales, representando la
idealidad de las mezclas lquidas.
Objetivo de la Unidad Temtica.
Capacitar al estudiante en: la determinacin de Propiedades termodinmicas para
mezclas de composicin variable comportamiento ideal y, resolver problemas de
sistemas de equilibrio liquido- vapor considerando comportamiento ideal.
3
J0 = IJP - SJI (1.11)
J 0) = (nI)JP - (nS)JI (2.1)
J(n0) = _
o(n0)
oI
2.1. RELACIN ENTRE PROPIEDADES PARA MEZCLAS HOMOGENEAS DE
COMPOSICIN VARIABLE.
En el tema 1, se estudio las relaciones entre propiedades para sustancias puras
(sistemas de composicin constante), en este apartado se determinan las
relaciones tomando en cuenta la variacin de la composicin. Para determinar
estas relaciones partamos de la ecuacin (1.11) del tema 1.
Matemticamente se establece la relacin directa:
G(f)=G(T,P)
La ecuacin (1.11) no establece relacin con el cambio de la composicin. La
ecuacin (1.11) se escribir tomando en cuenta los moles ya que al tratarse de
mezclas, estos variarn.
(n
Veamos como es Gibbs en funcin de T, P y los moles de los componentes
presentes.
G(f)= G(T,P, n
1
, n
2
,..n
i
)
_
P,n
JI +_
o(n0)
oP
_
1,n
JP +_
o(n0)
on
_
1,P,n]
Jn
(2.2)
J(n0) = -(nS)JI + (nI)JP +_
o(n0)
on
n
j
, representa todos los moles de cada componente presente en la mezcla con la
excepcin de n
i
.
Comparando (2.1) y (2.2) se puede escribir:
_
1 ,n]
Jn
(2.S)
,P
Slo falta definir el ltimo termino de la ecuacin (2.3), para ello se definir un
nuevo concepto el potencial qumico.
4
p
= _
o(n0)
on
Segn Gibbs, el potencial qumico p
coincide con el cambio que sufre la energa
libre de Gibbs cuando se aade una cantidad infinitesimal de moles de i a la
mezcla manteniendo constante la T, y P.
_
1,P,n]
(2.4)
p
= _
o(nu)
on
Si aplicramos el mismo procedimiento anterior a las ecuaciones fundamentales
restantes del tema 1, el potencial qumico queda tambin definido como:
_
v,S,n]
(2.S)
p
= _
o(nE)
on
_
P,S,n]
(2.6)
p
= _
o(nA)
on
_
v,1,n]
(2.7)
J(n0) = -(nS)JI + (nI)JP + p
Jn
(2.8)
La ecuacin (2.3) la podemos escribir como:
La ecuacin (2.8) representa el cambio de la energa libre de Gibbs, para
sistemas de composicin variable de fase homognea.
2.2. POTENCIAL QUMICO COMO CRITERIO DE EQUILIBRIO DE FASES.
Partamos de la ecuacin (1.12) de equilibrio que se estudio en el tema 1:
0
,
P T
dG
(1.12)
La desigualdad exige que todos los procesos irreversibles que se realizan a T y P
constante deben ocurrir en tal direccin de modo que la funcin total de Gibbs del
sistema disminuya. Puesto que la funcin de Gibbs disminuye en todos los
cambios hacia el equilibrio en un sistema a T y P constante, entonces el estado
de equilibrio para determinadas T y P debe ser aquel estado para el cual la
funcin de Gibbs tiene un valor mnimo con respecto a todas las variaciones
posibles a T y P dadas. En el estado de equilibrio la igualdad de la ecuacin
5
J0
1,P
= u (2.9)
y [
nH (nH)
u
+ (nH)
[
(2.1u)
0 = (n0)
u
+ (n0)
[
(2.11)
J(n0)
u
= -(nS)
u
JI +(nI)
u
JP +p
u
Jn
u
(2.12)
J(n0)
[
= -(nS)
[
JI + (nI)
[
JP + p
[
Jn
[
(2.1S)
J0 = p
u
Jn
u
+
[
Jn
[
(2.14)
(1.12) es vlida y esto significa que pueden ocurrir variaciones diferenciales en el
sistema a P y T constante sin producir un cambio de G. Por tanto, se establece un
criterio de equilibrio como:
La relacin (2.9) es la preferida sobre lo cual se basan los clculos de
equilibrio, debido a que es ms conveniente tratar T y P como constantes que
tratar con otras parejas de propiedades de estado.
La aplicacin de (2.9) al equilibrio de fases exige una expresin que
incorpore las composiciones o nmeros de moles de las especies qumicas en las
fases individuales. Se establece entonces que para cualquier propiedad
termodinmica la suma de sus funciones en las fases presentes representa la
propiedad total, es decir para 2 fases o por ejemplo se tiene:
=
Para Gibbs:
Aplicando (2.8) a ambas fases se tiene:
Sumando (2.12) y (2.13) para conseguir el cambio de G total del sistema, se
tiene:
Supongamos un sistema cerrado mostrado en la Fig 2.1, donde las propiedades
macroscpicas no muestran variacin de T y P la ecuacin (2.9) se cumple y la
ecuacin (2.14) queda:
Fig. 2.1. Equilibrio de dos fases coexistiendo a T y P fijas.
Fuente: www.lv-soft.com/
6
u = p
u
Jn
u
+
[
Jn
[
(2.1S)
Jn
u
= -Jn
[
(2.16)
u = p
u
Jn
u
-
[
Jn
u
(2.17)
u
p
u
= p
[
(2.18)
p
u
= p
[
= p
n
(2.19)
Donde:
La ganancia de moles de i en la fase o es proporcional a la perdida de moles de i
en la fase [. Sustituyendo (2.16) en (2.15):
Y que los Jn son cantidades independientes y arbitrarias la nica forma que
esta ecuacin pueda cumplirse es que:
Generalizando para mltiples fases:
Las ecuacin (2.19) representa el criterio de equilibrio para sistemas sin reaccin
qumica. Una diferencia en el potencial qumico de una especie particular
representa una fuerza impulsora para el transporte de esa especie, de
manera anloga a como la diferencias de temperatura y de presin representan
fuerzas impulsoras para la transferencia de calor y de momentum. Como un
principio general, cuando las fuerzas impulsoras se hacen cero, el proceso
de transporte cesa y se establece la condicin caracterizada por el trmico
equilibrio.
7
E
1,P
g
= y
1,P
k
(2.2u)
S
1,P
g
= y
(1,P)
- R y
ln
(2.21)
2.3. MEZCLAS DE GASES IDEALES: PROPIEDADES TERMODINMICAS DE
LAS MISMAS.
Para la determinacin de las propiedades en mezclas de gases ideales,
existen diferentes mtodos. Se deben mantener las mismas suposiciones que se
hacen cuando la sustancias es ideal, las cuales son: el volumen de la molcula es
infinita y no existen interacciones moleculares (se desprecian). Al tener estas
caractersticas Gibbs plantea en su teorema que: las propiedades termodinmicas
totales de una mezcla de gases ideales, son la suma de las propiedades totales
de los componentes individuales, evaluados a la T de la mezcla, pero a su presin
parcial.
En el apartado 2.4 pg. 123 de la gua de la Prof. Yolanda Reyes publicada
en el bloque del tema 2 del saln virtual, se establece de manera detallada la
deduccin de las ecuaciones para mezclas de gases ideales. En esta gua de
estudio se mostraran las ecuaciones para seguir un ordenamiento en el tema.
Una mezcla cuyos componentes son gases ideales y que se comportan
como gas ideal, su volumen total obedece a la ecuacin de estado de gas ideal.
Los volmenes de i en la mezcla sern igual a los de componentes puros a la
misma T y P, es decir que mantiene la propiedad. Cada componente de la mezcla
de gases ideales se comporta como si ocupara un volumen total, pero a la
presin parcial del componente.
Entalpa:
Entropa:
Mediante relaciones entre propiedades se consiguen las siguientes:
Energa libre de Gibbs
8
0
1,P
g
= y
(1,P)
+RI y
ln
(2.22)
p
g
= 0
g
(1,P)
+RI lny
(2.2S)
E
SI
= x
(2.24)
S
SI
= x
-R x
ln
x
(2.2S)
0
SI
= x
+ RI x
ln
x
(2.26)
p = 0
L
(1,P)
+ RI lnx
(2.27)
Potencial qumico
2.4. SOLUCIN IDEAL: CONCEPTO Y DETERMINACIN DE LAS
PROPIEDADES.
Para las soluciones lquidas ideales, se tiene la analoga aplicada al sistema
de mezcla de gases ideales. Para diferenciar la fase lquida de la gaseosa, se
utilizaran las ecuaciones anteriores pero la composicin se mostrara con x. Estas
soluciones son aquellas que poseen molculas de tamaos similares y las fuerzas
de interaccin molecular son semejantes. Los volmenes molares para las
soluciones ideales son aditivos. Las fuerzas de atraccin y repulsin entre
molculas al ser tan semejantes y ser vectores opuestos se anulan. Es por esta
razn que las ecuaciones para el clculo de propiedades son anlogas a las de
mezclas de gases ideales.
Entalpa:
Entropa:
Mediante relaciones entre propiedades se consiguen las siguientes:
Energa libre de Gibbs
Potencial qumico
SI
Las soluciones cuyas molculas son de tamaos semejantes y de la misma
naturaleza qumica con frecuencia se comportan del mismo modo que la solucin
ideal. Ejemplos de ellas son: mezclas de ismeros, tales como orto, meta y
paraxileno, las series homologas como n-hexano/n-heptano, estanol/propanol,
entre otros.
PROBLEMA 1
Un tanque rgido y aislado se divide
en 2 compartimientos y est
conectado por medio de un una
vlvula. Un compartimiento contiene
0,22 kgmol de gas oxigeno (0
2
) a 40
C y 100 kPa, y el otro compartimiento contiene 0,14 kgmol de Nitrgeno
(N
2
) a 20 C y 150 kPa. Despus se abre la vlvula y se deja que los gases
se mezclen. Determine la entropa de la mezcla.
Datos adicionales:
C
v
N
2
= 23,78 kJ/kgmol K
C
v
O
2
= 18,42 kJ/kgmol K
Observacin: Los C
v
y C
p
pueden buscarse en las diferentes literaturas de
termodinmica bsica o aplicada. En este caso el problema los trae como data
adicional, las presiones de los gases son bajas comparadas con sus condiciones
crticas, su comportamiento es ideal, y las condiciones no varan notablemente,
por ello los C
p
y C
v
se pueden asumir constantes en el proceso. En el caso de que
los C
v
sean funciones de la T, deben especificar una ecuacin matemtica que
permita obtener el valor. En el caso que no sea suministrado en el problema, el
se debe determinar primeramente si los gases son o no ideales, comparndolo
con sus condiciones crticas, si resulta un gas ideal puede tratarse como un
promedio de los C
v
a las diferentes temperaturas, mantenerlo constante (cuando
las temperaturas no varan mucho) u obtener los valores de esta data trmica en
literaturas donde se haya publicado tales como: el Manual del Ing. Qumico,
9
Himmelblau ( Pg. 661 sexta edicin), Smith (pag 684 sptima edicin), etc. En
donde aparecen los C
p
y C
v
como funciones de la T.
Anlisis del Problema:
Ambos gases inicialmente se encuentra como sustancias puras. Cuando se abre la
vlvula, los gases se mezclan, y dejan de tener sus propiedades iniciales para
formar una mezcla. Cada sustancia pasa a ser un componente de la mezcla. En el
caso de estudio estos gases se comportan como un sistema ideal, esto quiere
decir, que conservaran sus propiedades individuales basados en la Ley de Gases
ideales.
O
2
N
2
T
1
=40 C T
1
= 20 C
P
1
=100 kPa P
1
=150 kPa
10
Inicialmente
Apertura de la Vlvula
Finalmente
Fig. 2.2. Tanque rgido aislado problema 1. Arriba: estado inicial de la mezcla,
abajo: estado final de la mezcla. Fuente: Propia.
Ahora, tomaremos el sistema como el compartimiento completo, esto quiere decir
que el sistema no tendr salidas, ni entradas de masa, por lo tanto es un sistema
cerrado. La ecuacin de primera ley para un sistema cerrado se reduce en lo
siguiente:
U W Q = (2.28)
O
2
O
O
2
O
N
2
N
2
N
2
2
N
2
N
2
N
2
2
O
2
O N
2
N
2
N
2
2
O
2
O
2
N
2
N
2
N
2
N
2
O
O
2
N
2
O
2
N
2
O
2
N
2
O
2
N
2
O
2
N
2
O
2
N
2
O
2
N
2
Mezcla de O
2
y N
2
Temperatura de Mezcla = ?
Presin de Mezcla= ?
2
N
2
O
2
O
2
N
2
El sistema no presenta trabajo (por ser un sistema rgido), ni calor (sistema
aislado); por lo cual la ecuacin (2.28) queda reducida a:
11
0 U =
El cambio de energa interna est representado por la energa interna de cada
una de las sustancias presentes, esto quiere decir que el cambio de energa
interna del sistema ser la suma de las energas internas de los componentes de
la mezcla.
2 2 O N
U U U + = (2.29)
que a su vez es igual a cero
Podemos escribir entonces;
2 2
0
O N
U U + = (2.30)
Recordamos que el cambio de la energa interna lo podemos encontrar a travs
de los calores especficos a volumen constante, Cv.
T Cv U = (2.31)
Reescribimos nuevamente la ecuacin (2.30) al sustituir la ecuacin (2.31) en
ella y nos queda:
) ( ) ( 0
2 2
T Cv T Cv
O N
+ = (2.32)
Donde:
1 2
T T T = (2.33)
Ya que solo existen el estado inicial (1) y el estado final (2).
Sustituimos (2.33) en (2.32),
) ) ( ( ) ( 0
2 1 2 2 2 1 2 2 O O N N
T T Cv T T Cv + = (2.34)
Tenemos las temperaturas iniciales de cada uno de los compuestos, estado en el
cual no se ha abierto la vlvula, pero no tenemos la temperatura 2, la cual es la
temperatura de la mezcla, del estado final, cuando ya se ha abierto la vlvula.
Por ello la ecuacin (2.34), sirve para determinar la temperatura 2.
Sustituyendo los valores en la ecuacin (2.34), nos queda:
) ) 313 ( 42 , 18 ( ) 293 ( 78 , 23 0
2 2 2 2 O N
T
K kgmol
kJ
K T
K kgmol
kJ
=
Despejando la T
2
nos queda:
T2 = 301,73 K
La entropa de la mezcla se calcula por la ecuacin (2.21),
S
1,P
g
= y
(1,P)
- R y
ln
(2.21)
Debemos calcular la entropa S
i
, que es la entropa de cada uno de los
componentes cuando estn puros. La ecuacin (1.24) fue vista en el tema
anterior para determinar las S
i
, esta es la indicada debido a que la data
suministrada es T y P, para un gas ideal la ecuacin (1.24) se convierte en la
(1.51) tambin vista en el tema 1.
12
P
dP
R
T
dT
Cp dS
gi
=
T R n V P
(1.51)
Cuando integramos dichas ecuacin necesitaremos, la T
2
(ya calculada), y las
presiones parciales de cada uno de los componentes en el estado 2.
Observacin: la presin parcial de cada uno de los componentes es la que
se utiliza en la evaluacin del cambio de entropa en los estado 1 y 2, NO
la presin de mezcla Pm.
Para determinar las presiones parciales en el estado 2, debemos buscar la
Presin de la mezcla, para ello usaremos la ecuacin de los gases ideales:
=
(2.22)
Se debe calcular los moles de la mezcla, y el volumen de la mezcla. Para los
moles de mezcla, simplemente sumamos las cantidades dadas en el problema, ya
que es un sistema cerrado.
2 2 N O
n n n + =
kgmol n 36 , 0 =
El volumen de la mezcla, debemos buscarlo, sumando los volmenes de los
componentes individuales, esto quiere decir que los volmenes en el caso de
mezcla de gases ideales, son aditivos. Para encontrar estos volmenes de los
componentes, volvemos aplicar la ecuacin de gas ideal, a las condiciones
iniciales de cada uno de ellos.
2
2 2
02
O
O O
P
T R n
V
=
3
3
02
73 , 5
100
313 314 , 8 22 , 0
m
kPa
K
K kgmol
m kPa
kgmol
V =
=
Para el N
2
de igual manera pero a sus propias condiciones iniciales
3
2
27 , 2 m V
N
=
El volumen de la mezcla entonces ser:
2 2 N O
V V V + = = 8 m
3
Ahora si podemos buscar la Pm (presin de la mezcla)
V
n T R
Pm
=
3
8
36 , 0 73 , 301 314 , 8
m
kgmol K
K kgmol
kJ
Pm
=
kPa Pm 89 , 112 =
Se determinan ahora las presiones parciales de cada uno de los componentes en
el estado 2. Para ello necesitamos las fracciones molares de cada uno de los
componentes.
n
n
y
i
i
=
y
O2
= 0,61
y
N2
= 0,39
Las presiones parciales sern: Pm yi Pi = entonces tenemos
P
O2
= 68,86 kPa
P
N2
= 44,03 kPa
13
Busquemos ahora las entropas de cada una de las sustancias por la ecuacin
(1.51).
14
=
100
86 , 68
ln 314 , 8
313
73 , 301
ln 38 , 29
2
K kgmol
kJ
K kgmol
kJ
S
O
K kgmol
kJ
S
O
= 0246 , 2
2
De igual manera para el N
2
K kgmol
kJ
S
N
= 05 , 11
2
Finalmente aplicamos la ecuacin (2.21), y encontramos la entalpa de la mezcla.
K kgmol
kJ
S
gi
= 10 , 11
2.5. LEY DE RAOULT. DEDUCCIN A PARTIR DEL CRITERIO DE
EQUILIBRIO DE FASES.
La ley de Raoult es una ecuacin que permite relacionar la fase lquida con la fase
de vapor. Es la relacin ms simple para resolver problemas de equilibrio de
fases. Esta ecuacin se consigue a partir del criterio de equilibrio de fases
obtenido:
p
u
= p
[
= p
n
(2.19)
p
= p
L
(2.28)
0
g
(1,P)
+ RI lny
= 0
L
( )
+ RI lnx
(2.29)
RI ln
y
x
Supongamos dos fases en coexistencia, en nuestro caso nos interesa el estudio
lquido/vapor.
Si suponemos la fase de vapor como una mezcla de gases ideales, y la fase
lquida como una solucin ideal, las ecuaciones (2.23) y (2.27) se pueden
sustituir en (2.28)
1,P
Reagrupando en (2.29) trminos semejantes:
= 0
L
(1,P)
-0
g
(1,P)
(2.Su)
15
RI ln
y
Asumiendo que el lquido es incompresible, Gibbs no depende de la P solo de la
T, quedando (2.30) como:
= 0
L
(1,P
i
sct
)
-0
g
(1,P)
(2.S1)
g
(1,P)
J0 = IJP - SJI (1.11)
J0
= I
JP (2.S2)
0
g
(1,P
i
sct
)
- 0
g
,P)
= RI ln
P
sut
P
Se obtiene la expresin de 0 mediante la ecuacin fundamental vista en el
tema 1 para sustancias puras (1.11).
A T constante;
Integrando desde la presin de saturacin hasta la P del sistema y sustituyendo
la ecuacin de gas ideal en Vi;
(1
(2.SS)
RI ln
y
Despejando en (2.33) el trmino buscado, se sustituye en (2.31)
= 0
L
(1,P
i
sct
)
- 0
g
(1,P
i
sct
)
+RI ln
P
sut
P
(2.S4)
y
En el equilibrio para una sustancia pura el cambio de la energa libre de Gibbs es
cero, la ecuacin (2.34) queda:
=
P
sut
P
(2.SS)
La ecuacin (2.35) es la conocida como la Ley de Raoult.
La constante de Equilibrio (K
I
):
Una medida apropiada de esta tendencia con respecto al equilibrio V-L viene
dada por la siguiente relacin:
xi
yi
K
i
=
(2.36)
K
i
: generalmente se le denomina coeficiente de reparto o constante de equilibrio.
Si K
i
< 1 quiere decir que el componente (i) se concentra en la fase lquida y se
considera entonces como pesado y la fase lquida es rica en este componente. Si
K
i
> 1 quiere decir que el componente (i) tiende a concentrarse en la fase de
vapor y se dice que el componente es liviano, y la fase de vapor es rica en este
componente.
De la ecuacin (2.35) se tiene:
16
sat
i
P P
i i
x y =
quiere decir que,
P
Pi
K
sat
i
=
(2.37)
2.6. CONSTRUCCIN DE DIAGRAMA P vs. x,y; T vs. x, y PARA SISTEMAS
BINARIOS UTILIZANDO LA LEY DE RAOULT.
A
C
x
1
, y
1
A C
17
x
1
, y
1
x
1
z
1
y
1
0 1
(Temp
T
eratura)
T vs. x1
T vs. y1
b) DIAGRAMA T vs. x
1
, y
1
, a Presin Constante
Vapor Sobrecalentado
Lquido Subenfriado
Curva de Roco
Curva de Burbuja
T
1
sat
T
2
sat
L-V
B
P
2
sat
a) DIAGRAMA P vs. x
1
, y
1
a Temperatura Constante
0 1
P vs. x1
P vs. y1
Vapor Sobrecalentado
Lquido Subenfriado
Curva de Burbuja
Curva de
Roco
L-V
P
1
sat
P
n) (Presi
x
1
z
1
y
1
A B
C
Fig. 2.3. Diagramas. Sistemas Binarios a) Presin vs. x1/y1 y b) Temperatura
vs. Composicin. Fuente: Propia.
Regla de la palanca:
Es una regla matemtica vlida para cualquier diagrama binario. En estos
sistemas, se utiliza una regla que permite establecer la relacin entre la cantidad
de masa contenida en cada una de las fases en equilibrio.
En regiones de una sola fase, la cantidad de la fase pura es 100% o x=1. En
regiones bifsicas, sin embargo, se deber calcular la cantidad de cada fase. Una
tcnica es hacer un balance de materiales. Por ejemplo: una mezcla de fraccin
molar (z) a Temperatura T y Presin P (Punto B) en los diagramas T x,y o
P x,y, forma un lquido que contiene una fraccin molar x (punto A) y un vapor
de fraccin molar y (punto C) (ver diagramas a y b) .
La mezcla contiene F (moles) que tiene una fraccin molar z
1
del componente
(1), la cual se separa en L (moles de lquido) y V (moles de vapor). Las
fracciones de cada una de las corrientes molares estn definidas para el lquido
como x
1
y para el vapor como y
1
.
El balance por componente para el componente 1 queda:
V y L x n z + =
1 1 1
(2.38)
El balance global:
V L F + =
(2.39)
Sustituyendo (2.39) en (2.38) se tiene:
V y L x V z L z + = +
1 1 1 1
(2.40)
Rearreglando queda finalmente;
AB
BC
x z
z y
V
L
=
=
) (
) (
1 1
1 1
(2.41)
La ecuacin (2.41) representa la ecuacin matemtica de la regla de la
palanca.
Para calcular las cantidades de lquido y de vapor, en digrama T-xy o P-xy, se
construye una palanca (lnea verde) sobre la isoterma o isobara con su punto de
18
apoyo en la composicin original de la mezcla (punto dado z
1
). El brazo de la
palanca, opuesto a la composicin de la fase cuya cantidad se calcula se divide
por la longitud total de la palanca, para obtener la cantidad de dicha fase.
Esta regla, establece que el
nmero de moles de las fases
en equilibrio es inversamente
proporcional a la relacin de
las longitudes de los
correspondientes segmentos
de las lneas de enlace.
19
PROBLEMA 2
El siguiente sistema benceno (1)/ etilbenceno (2) concuerda bastante
bien con la ley de Raoult.
Las presiones de vapor de los componentes puros vienen dada por la ecuacin de
Antoine (1.59) estudiada en tema 1.:
) (
) ( ln
K T C
B
A kPa P
sat
+
=
Las constantes de estos compuestos, las buscamos en el anexo 1 de esta guia,
las cuales son:
Sustancia A B C
Benceno (1) 13,8858 2788,51 -52,36
Etilbenceno(2) 14,0045 3279,47 -59,95
Fig. 2.4. Regla de la palanca para Diagramas
Binarios Temperatura vs. x1-y1. Fuente: Propia.
sa
t
0
Isoterma
T
2
B
1
A C
z
1
Se mide con una
regla la longitud
sa
t
T
1
T vs. y1
a) Prepare una grafica que muestre P en funcin de x1 y P en funcin de
y1, Para la temperatura de 90 C.
Solucin:
Con la Temperatura de 90 C calculamos las Presiones de saturacin de
ambos componentes. Estas Presiones sern los lmites en el rango de la
presin.
La temperatura se lleva a Kelvin, ya que la formula de Antoine esta expresada en
esta escala.
)
) ( 363 36 , 52
51 , 2788
8858 , 13 exp(
1
K
P
sat
+
=
kPa P
Sat
52 , 135
1
=
De modo similar para el etilbenceno, pero con las constantes de el.
kPa P
Sat
12 , 24
2
=
Asumimos los valores de las composiciones de la fase liquida, es decir
tomaremos entre 0 y 1, valores arbitrarios de x1.
x1
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
Para calcular las diferentes presiones del sistema para cada una de las
composiciones, utilizaremos la ley de Raoult (2.35), colocando la ecuacin
en funcin solo de la fase liquida (ya que son los valores de las
compasiones que se estan asumiendo) nos queda:
20
21
(2.42)
sat sat
sat
sat
P x P x P
P x P y
P x P y
2 2 1
2
1
1
2 2
1 1
+ =
=
=
Como:
1 1 2
1 2 1
x x
x x
=
= +
Sustituyendo en (2.42), y rearreglando nos queda;
( )
sat sat sat
P P P x P
2 2 1
1 + = (2.43)
Con esta ecuacin (2.43), determinamos las P para cada una de las
composiciones asumidas.
x1 P
0 24,1178541
0,1 35,2583354
0,2 46,3988168
0,3 57,5392981
0,4 68,6797794
0,5 79,8202607
0,6 90,9607421
0,7 102,101223
0,8 113,241705
0,9 124,382186
1 135,522667
Determinamos cada una de las yi, con la ecuacin (2.35) de Raoult.
P
P x
y
sat
1 1
1
=
x1 P y1
0 24,1178541 0
0,1 35,2583354 0,38437058
0,2 46,3988168 0,58416433
0,3 57,5392981 0,70659187
0,4 68,6797794 0,7893017
0,5 79,8202607 0,84892398
0,6 90,9607421 0,8939417
0,7 102,101223 0,92913546
0,8 113,241705 0,95740464
0,9 124,382186 0,98060988
1 135,522667 1
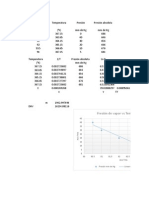
Y finalmente graficamos en papel milimetrado, las P en el eje de las Y, y
las composiciones x1, y1, en el eje de las X. En la fig. 2.5 se muestra.
DIAGRAMA BENCENO/ ETILBENCENO P VS. X1,
Y1, A T=90C
0
20
40
60
80
100
120
140
160
0 0,2 0,4 0,6 0,8 1
x1, y1
P
(
k
P
a
)
P vs. x1
P vs. y1
Fig. 2.5. Diagrama de equilibrio L-V (P-xy) para el sistema Benceno-etilbenceno
a 90C, asumiendo validez de la ecuacin de Raoult. Fuente: Propia.
b) Prepare una grafica que muestre T en funcin de x1 y T en funcin de
y1, Para la presin de 90 kPa.
Calculamos la T1 y T2 de saturacin por la ecuacin de Antoine a la Presin
de 90 kPa. Estas sern los lmites del Diagrama.
T1sat= 349,45 K
T2sat= 404,98 K
Asumimos las T del sistema entre las temperaturas de saturacin de de los
componentes. (estos valores son arbitrarios, pueden seleccionar otros).
22
T (K)
349,45
365
370
375
380
385
390
395
404,99
T
1
sat
T
2
sat
Con cada temperatura asumida calculamos la P1 y P2 de saturacin.
T (K) P
1
sat
P
2
sat
349,45 90 14,5350153
365 143,532834 25,8911636
370 165,167865 30,7924805
375 189,238717 36,4206677
380 215,91912 42,8522136
385 245,386233 50,1678935
390 277,820169 58,4526847
395 313,403523 67,7956619
404,99 394,64348 90
Con la ecuacin (2.43), despejamos x1 y las calculamos.
sat sat
sat
P P
P P
x
2 1
2
1
=
23
T (K) P1sat P2sat x1
349,45 90 14,5350153 1
365 143,532834 25,8911636 0,544950069
370 165,167865 30,7924805 0,440612837
375 189,238717 36,4206677 0,350608664
380 215,91912 42,8522136 0,272425198
385 245,386233 50,1678935 0,204038753
390 277,820169 58,4526847 0,143810353
395 313,403523 67,7956619 0,090405649
404,99 394,64348 90 0
Por medio de la ecuacin de Raoult calculamos las y1.
P
P x
y
sat
1 1
1
=
T K P1sat P2sat x1 y1
349,45 90 14,5350153 1 1
365 143,532834 25,8911636 0,544950069 0,86909142
370 165,167865 30,7924805 0,440612837 0,80861202
375 189,238717 36,4206677 0,350608664 0,73720816
380 215,91912 42,8522136 0,272425198 0,65357566
385 245,386233 50,1678935 0,204038753 0,55631445
390 277,820169 58,4526847 0,143810353 0,44392685
395 313,403523 67,7956619 0,090405649 0,3148161
404,99 394,64348 90 0 0
Finalmente construimos el diagrama. En la Fig. 2.6 se muestra.
24
DIAGRAMA BENCENO/ETILBENCENO T VS. x1, y1,
A LA P=90 kPa
340,00
350,00
360,00
370,00
380,00
390,00
400,00
410,00
0 0,2 0,4 0,6 0,8 1
x1, y1
T
(
K
)
T vs. x1
T vs. y1
Fig. 2.6. Diagrama de equilibrio L-V (T-xy) para el sistema Benceno-etilbenceno
a 90 kPa, asumiendo validez de la ecuacin de Raoult. Fuente: Propia.
2.7. CLCULO DE PUNTO DE BURBUJA, PUNTO DE ROCO Y
VAPORIZACIN INSTANTANEA.
Punto de Burbuja: Se define como la P o la T a la cual se forma la primera
gota de vapor cuando un lquido se dilata (P = P
B
) o se calienta (T = T
B
) a T
o P = constante respectivamente. La ecuacin para multicomponentes viene de
generalizar la ecuacin (2.42).
B
i
sat
i i
P P P x = =
(2.44)
Presin de Roco: Se define como la P o la T a la cual se forma la primera
gota de roco cuando un vapor se comprime (P = PR) o se enfra (T = TR) a T o
25
P cte respectivamente. Su ecuacin se consigue de modo anlogo a la ecuacin
(2.42). Se parte de la ecuacin (2.35).
y
P = x
sut
(2.35)
26
x
1
=
y
1
P
P
1
sut
Apliquemos la ecuacin (2.35) para 2 componentes y finalmente se generaliza
para multicomponentes, se despejan las xi para que la ecuacin quede en funcin
de yi:
x
2
=
y
2
P
P
2
sut
1 =
y
1
P
P
1
sut
+
y
2
P
P
2
sut
Se suman:
P
R
=
1
y
1
P
1
sut
Se despeja la presin, que sera la presin en funcin de la composicin de la
fase de vapor (presin de roco).
(2.4S)
+
y
2
P
2
sut
Generalizando:
= =
i
sat
i
i
R
P
Y
P P
1
(2.46)
Si se especifica la composicin de la fase liquida (x1, x2, x3, xi......xn) y T o P,
se conoce como punto de burbuja.
Si se especifica la composicin de la fase vapor (y1, y2, y3, yi yn) y T o P,
entonces se conoce como punto de roco.
Procedimientos:
Tabla 2.1. Procedimientos algortmicos para el clculo de Puntos de Burbuja y
Roco. Fuente: Propia.
A) Calculo de Presin de Burbuja. Dadas
la T y Composiciones de la fase liquida
(xi)
1. Calculo de Pi
sat
por Antoine u otro
mtodo.
2. Calculo de la Presin por
= xi Pi P
sat
3. Calculo de yi por Raoult.
4. FIN
C) Calculo de Temp de Burbuja. Dada la P
y las composiciones de la fase liquida (
xi)
1. Asumir la T del sistema
2. Calculo de Pi
sat
por Antoine, con la T
asumida.
3. Calculo de las yi por Raoult
4. Se debe cumplir que la
=1 yi
5. Si la temperatura que fue
asumida
1 yi
en paso 1 esta errada, por lo tanto, se asume
otra
temperatura. Este paso se repite hasta que se
cumpla 4.
6. Cuando se cumpla 4, FIN
B) Calculo de Presin de Roco. Dadas la T
y las composiciones de la fase de vapor (
yi).
1. Calculo de Pi
sat
por Antoine u otro
mtodo.
2. Calculo de la Presin por
=
sat
Pi
yi
P
1
3. Calculo de xi por Raoult.
sat
Pi
P yi
xi
=
4. FIN
D) Calculo de Temp de Roco. Dada la P y
las composiciones de la fase de vapor (
yi).
1. Asumir la T del sistema
2. Calculo de Pi
sat
por Antoine, con la T
asumida.
3. Calculo de las xi por Raoult
4. Se debe cumplir que la
=1 xi
5. Si
1 xi la temperatura que fue
asumida
en paso 1 esta errada, por lo tanto, se asume
otra
Temperatura. Este paso se repite hasta que se
cumpla 4.
6. Cuando se cumpla 4, FIN
PROBLEMA 3.
La presin sobre un gas licuado de petrleo de composicin mostrada en
la tabla, se mantiene en 4053 kPa.
Componente Etano Propano n-Butano
%Molar 15 40 45
27
a) Calcular la temperatura de burbuja
b) Calcular la temperatura de roco.
c) Explique con sus palabras, para que se calculan ambas temperaturas.
Solucin:
Calculo de Temperatura de Burbuja
a) Para la solucin de este problema se seguir el procedimiento C, ilustrado en
la tabla 2.1.
1. Se asume una T. Para evitar tantas iteraciones, se realizara un promedio
ponderado de las temperaturas de saturacin de los componentes; el cual
permitir acercarse a un valor prximo.
Las temperaturas de saturacin se buscan por Antoine, las cuales son:
T1sat= 300,1186395 K
T2sat= 371,6688225 K
T3sat= 436,7547797 K
La Temperatura que se asumir ser:
= xi Ti T
sat
K T 22 , 390 =
2. Calculo de Pi
sat
con T asumida.
P1sat 14724,78428 kPa
P2sat 5333,781586 kPa
P3sat 2011,644023 kPa
3. Calculo de yi por Raout
P
P x
y
sat
1 1
1
=
y1 0,54495871
y2 0,52640331
y3 0,22335056
28
4.
= 2947 , 1 yi
No se cumple el paso 4.
5. Asumimos otra temperatura T= 371 K, podemos ver que la nueva
temperatura asumida est por debajo de la primera, esto se debe a que se
necesita disminuir las presiones de saturacin para que las composiciones
disminuyan de acuerdo a la ley de Raoult.
P1sat 11816,13637 kPa
P2sat 4010,864978 kPa
P3sat 1423,4785 kPa
y1 0,43731075
y2 0,3958416
y3 0,15804721
= 9911 , 0 yi
Un valor bastante aproximado a la unidad, si se quiere seguir iterando, se puede
usar la tcnica de interpolacin con este valor, y otra por arriba de este.
6. El valor final de
T
B
= 371,6 K
P1sat 11901,88622 kPa
P2sat 4048,65145 kPa
P3sat 1439,788555 kPa
y1 0,44048432
y2 0,39957083
y3 0,15985809
= 9999 , 0 yi
29
Calculo de Temperatura de Roco
b) Para este clculo se utilizara el procedimiento D de la tabla 2.1.
1. Temperatura asumida, igual a la de la letra a.
K T 22 , 390 =
2. Igual a letra a.
3. Calculo de xi por Raoult.
sat
Pi
P yi
xi
=
x1 0,04128753
x2 0,30394945
x3 0,90664649
4.
= 2518 , 1 xi
5. Asumimos otra temperatura, debemos aumentar las presiones de saturacin
para disminuir las fracciones de xi, para ello debemos aumentar la temperatura. L
nueva temperatura que asumiremos es de 405 K
P1sat 17183,08263 kPa
P2sat 6511,974959 kPa
P3sat 2561,277036 kPa
x1 0,03538073
x2 0,24895673
x3 0,71208619
= 996 , 0 xi
6. La
T
R
= 405 K
Ya que la sumatoria de las xi es muy cercana a 1.
30
PROBLEMA 4. (Ejercicio resuelto en Smith Van Ness Cuarta Edicin pg.
320 capitulo 10).
Para un sistema acetona (1) / acetonitrilo (2) / Nitrometano (3), se
tienen las siguientes ecuaciones de Antoine:
31
237.22 T
2940.46
14.5463 P Ln
Sat
1
+
=
224.00 T
2945.47
14.2724 P Ln
Sat
2
+
=
209.00 T
2972.64
14.2043 P Ln
Sat
3
+
=
Donde T esta en C, y la presin de vapor en kPa. Considerando que la ley de
Raoult es apropiada para el sistema, calcule:
a) P y {y
k
}, dado T = 80 C, x
1
= 0.25, x
2
= 0.35, x
3
= 0.40
b) P y {x
k
} dado T = 70 C, y
1
= 0.50, y
2
= 0.30, y
3
= 0.20
c) T y {x
k
}, dado P = 80 kPa, x
1
= 0.30, x
2
= 0.45, x
3
= 0.25
d) T y {x
k
} dado P = 90 kPa, y
1
= 0.60, y
2
= 0.20, y
3
= 0.20
Solucin:
a) Presin de Burbuja procedimiento A de la tabla 2.1:
1. Se evalan las C 80 P
Sat
i
usando las Relaciones de Antoine del Problema:
kPa 195.75 P
Sat
1
=
kPa 97.84 P
Sat
2
=
kPa 50.32 P
Sat
3
=
2. ( ) ( ) ( ) ( ) ( ) ( ) 50.32 0.40 97.84 0.35 195.75 0.25 P + + = kPa 103.31 P = = P
B
3.
P
P X
Y
Sat
i i
i
=
y
1
= 0.4737
y
2
= 0.3315
y
3
= 0.1948
b) Presin de Roco. Procedimiento B de la tabla 2.1.
C 70
Sat
i
P por Antoine: Se evalan
kPa 43.80 P 70.37; P 144.77; P
Sat
3
Sat
2
Sat
1
= = =
Se calcula la presin de roco dada la composicin de la fase de vapor por la
ecuacin (2.46):
=
sat
Pi
yi
P
1
P
R
= 74,27 kPa
Finalmente se calculan las xi por Raoult
x
1
= 0.2569
x
2
= 0.3166
x
3
= 0.4269
c) Temperatura de Burbuja. Procedimiento C de la tabla 2.1.
Las temperaturas de saturacin se buscan por Antoine, las cuales son:
T1sat= 52,07 C
T2sat= 73,81 C
T3sat= 93,64 C
La Temperatura que se asumir ser:
= xi Ti
sat
T
C T 25 , 72
0
=
32
Se calculan las presiones de saturacin a esa temperatura, se comprueba que la
sumatorio de las fracciones de vapor de igual a uno. Si no, se vara la
temperatura hasta cumplir con la condicin de burbuja.
La temperatura de burbuja ser entonces
33
C T 60 , 68 =
y1 0,5196
y2 0,3773
y3 0,1031
d) Temperatura de Roco. Procedimiento D de la tabla 2.1.
Se sigue el procedimiento del problema 3, parte b
A la presin de 90 kPa
T1sat= 55,47 C
T2sat= 77,40 C
T3sat= 97,32 C
C T 23 , 68
0
=
Con la T
0
asumida, se comprueba que la sumatoria de las xi sean igual a 1, sino
se sigue iteracin. Los resultados de este son:
C T 95 , 73 =
x1 0,3303
x2 0,2240
x3 0,4457
Calculo de vaporizacin instantnea. Comportamiento ideal. Separacin
instantnea.
Una mezcla de componentes en determinada
fase (lquida o gaseosa), segn sean los valores
de la temperatura y la presin (T
1
,P
1
), y cuya
composicin molar es z
i
, se quiere llevar a la
regin difsica, para separar posteriormente las
corrientes de lquido, que tiene una composicin
x
i
, y de vapor, cuya composicin es y
i
.
En el caso de que la mezcla inicial se encuentre en fase lquida, el vapor formado
despus de la vaporizacin instantnea recibe el nombre de fraccin de lquido
evaporado, y la fraccin de la mezcla que permanece en fase lquida recibe el
nombre de lquido remanente, mientras que si la mezcla inicial se encuentra en
fase gaseosa, el producto en fase lquida y en fase gaseosa sern la fraccin de
vapor condensado y el vapor remanente. En cualquier caso, el producto en fase
lquida y en fase gaseosa sern designados en esta gua con las letras L y V
respectivamente, y la mezcla de entrada a la cmara con la letra F.
34
Descripcin del Proceso.
El proceso ms simple de destilacin
continua, es el de vaporizacin
instantnea de equilibrio de etapa
simple adiabtica, que se representa
en la figura 2.7. La temperatura de
la corriente ascendente y la cada de
presin en la vlvula se ajustan para
vaporizar la alimentacin hasta el
punto deseado, mientras que el
tambor proporciona espacio de
Fig.2.7. Equipo de Separacin en la Vaporizacin
Instantnea de Equilibrio. Fuente: Medina W.
35
desenganche para permitir que se separe el vapor del lquido.
Un flash es una destilacin en una sola etapa, en la cual se vaporiza
parcialmente una corriente de alimentacin para obtener un vapor y un lquido en
equilibrio. El vapor est constituido principalmente por los componentes ms
voltiles. En la figura 2.7, se precalienta la alimentacin lquida F bajo presin, y
se vaporiza parcialmente cuando la presin se reduce en la vlvula de expansin
(proceso adiabtico). Posteriormente, las fases vapor y lquido en equilibrio se
separan en el tambor. Si la vlvula se elimina, el proceso se lleva a cabo
calentando el lquido a baja presin, vaporizndolo parcialmente y separando las
fases en el tambor.
En el esquema mostrado en la figura 2.7, la temperatura del tambor se
mantiene constante mediante control del caudal del medio de calefaccin (vapor
de Agua), la presin del recipiente controla la salida del producto en fase vapor, y
el caudal de lquido lo determina un control de nivel. Obviamente, tambin se
puede utilizar otro esquema de control.
La importancia del estudio de la destilacin flash se debe fundamentalmente a:
- Este proceso se utiliza para preparar corrientes de alimentacin para
procesamientos subsiguientes.
- Los clculos flash se utilizan ampliamente para determinar la condicin
termodinmica de una corriente de composicin, presin y temperatura
conocida.
- Los mtodos computacionales utilizados para calcular una etapa de equilibrio
son de fundamental importancia, de hecho, se puede disear una columna de
destilacin por extensin de los mtodos desarrollados para los clculos flash.
El clculo de vaporizacin instantnea consiste en la determinacin de las
composiciones yi, xi y cantidades de las fases lquidas y vapor (L y V) en
equilibrio a partir de la P y T y composicin zi de la corriente de alimentacin.
El proceso puede realizarse continuamente si un lquido (F) se estrangula a
travs de una vlvula de expansin en un tanque a una P dada producindose
una separacin de ambas fases lquida (L) y vapor (V) a una P y T particulares,
donde las corrientes del proceso F, L y V se pueden expresar por unidad de masa
o mol.
Desarrollo de una de las expresiones para el clculo de vaporizacin
instantnea. Calculo de: L, V, x
i
, y
i
Balance Global para el separador de la
Fig. 2.8:
F = L + V (2.47)
Considerando Base de clculo:
F = 1mol de mezcla
Composicin de la alimentacin= z
i
Se despeja L de la ecuacin (2.47) para
colocar la ecuacin en funcin de V.
36
1 - I
1 = V + L
I =
(2.48)
Fig.2.8. Balance de materiales para el separador.
Fuente: Propia
Balance de componente:
V + =
i i i
y L x z
(2.49)
Sustituyendo (2.48) en (2.49)
( ) V 1
i i i
y V x z + = (2.50)
Se incluye la definicin de la constante de equilibrio mediante la ecuacin (2.36),
y despejando de ella la composicin de la fase lquida:
x
=
y
(2.S1)
Se sustituye (2.51) en (2.50)
( ) V yi V
ki
yi
zi + = 1
ki
ki V yi V yi
zi
+
=
) 1 (
ki V yi V yi ki zi + = ) 1 (
ki V yi V yi yi ki zi + =
Se saca factor comn
( ) ki V V yi ki zi + = 1
[ ] ) 1 ( 1 + = ki V yi ki zi
Despejando y
i
) 1 ( 1 +
=
ki V
ki zi
yi
(2.52)
Como
=1
i
y
) ki ( V
ki zi
1 1
1
+
=
(2.53)
Con esta ecuacin se determina V por ensayo y error hasta que se cumpla la
condicin. Para determinar la corriente molar liquida (L), se hace un balance
sencillo de masa con la ecuacin (2.48). Los K
i
se obtienen de la ecuacin (2.37):
P
P
K
sat
i
i
=
(2.37)
Luego como
xi
yi
K
i
=
ki
yi
x
i
=
37
Otros Mtodos de clculo en Vaporizacin Instantnea.
El clculo de un punto sobre la regin equilibrio de fases, que se encuentra entre
el punto de roco y el punto de burbuja, se conoce como clculo de una
vaporizacin instantnea isotrmica, porque se especifica T. Excepto para una
mezcla binaria ideal, los procedimientos para calcular una vaporizacin
instantnea isotrmica son iterativos. Un mtodo popular es el propuesto por
Rachford y Rice [J. Pet. Technol., 40 (10), Sec.1,p.19, y Sec.2,p.3 (October,
1952)], en el que se toma:
a) F = 1.0 mol de mezcla a la entrada de la cmara.
b) El balance en moles del componente i: z F y V x L
i i i
= + (2.49)
c) La relacin de la distribucin de fases: K
y
x
i
i
i
= (2.36)
d) El balance total de moles: F L V = + (2.47)
Al combinar los incisos (a), (b), (c) y (d), se obtiene:
1) V(K 1
z
x
i
i
i
+
=
De la ecuacin (2.36): y
i
(2.54) K x
i i
=
y, siendo F = 1.0 moles, la ecuacin (2.47) proporciona: L 1 V = (2.48)
Al sustituir las expresiones (2.54) y (2.48) en la ecuacin (2.49):
V) (1 x V x K z
i i i i
+ =
V] 1 V [K x z
i i i
+ =
1)] V(K + [1 x z
i i i
=
finalmente, despejando x
i
:
1) V(K 1
z
x
i
i
i
+
= (2.56)
38
Con la ecuacin (2.56) y (2.52) se determina la ecuacin de Rachford, J. y Rice,
J. proveniente de:
39
y 1 y x 1
i i
= Sabiendo que: =
se define: f {V} y x 0
i i
= =
y, a partir de las ecuaciones (2.56) y (2.52):
f {V}
K z
1 V(K 1)
z
1 V(K 1)
0
i i
i
i
i
=
+
+
=
f {V}
z (K 1)
1 V(K 1)
0
i i
i
=
+
=
(2.57)
La ecuacin (2.57) se aplica para hallar una de las tres variables siguientes: P, V
T, siempre que se conozca el valor de las dos restantes y la composicin de la
alimentacin, z
i
. Tambin se aplica, luego de algunas simplificaciones, en la
determinacin de la temperatura y presin de burbuja y roco. La ecuacin (2.57)
sirve para clculos ms complicados donde la constante de equilibrio no solo
dependa de T y P sino tambin de las composiciones de ambas fases. En el tema
4 apartado 4.7, se ver como la ecuacin (2.57) puede resolverse mediante uso
de herramientas matemticas sin necesidad de tantear.
PROBLEMA 5.
La corriente de vapor de una columna fraccionadora que pasa al
condensador tiene el siguiente anlisis: etano 15%, propano 20%, i-
butano 60% y n-butano 5%. Si la corriente que est en el condensador
esta a 300 K y 730 kPa, cul es la composicin de la fase lquida y
gaseosa, y cules son los flujos molares de liquido y vapor?
Solucin:
Antes de hacer el clculo de evaporizacin instantnea se debe comprobar para
las condiciones establecidas, que el sistema est en presencia de ambas fases
lquida y vapor, la regin difsica. Si la P est entre P
B
y P
R
el sistema es difsico.
Las presiones de vapor para cada componente a 300 K
( ) kPa , P , P ; , P 4047,75; P
Sat
4
Sat
3
Sat
2
Sat
1
047 257 087 367 38 990 = = = =
Se calcula la presin de burbuja y de roco, para establecer si el sistema est o
no en equilibrio de fases a esas condiciones de T y P. Es importante destacar que
tambin puede establecerse por las Temperaturas de burbuja y roco, sin
embargo por tratarse de clculos con mayor grado de dificultad ya que se tiene
que tantear, es preferible utilizar como mtodo las presiones de burbuja y roco.
Condicin del Punto de Burbuja
=
Sat
i i
P X P (2.44) P = P
B
= 1 Y
i
Sat
4 4
Sat
3 3
Sat
2 2
Sat
1 1 B
P X P X P X P X P + + + =
Las composiciones son tomadas del mismo enunciado, asumiendo una base de
clculo de 100 moles se tiene entonces que las composiciones son:
( ) ( ) ( ) ( ) ( ) ) , )( , ( 367,087 0.60 990,38 0,20 4047,75 0.15 P
B
047 257 05 0 + + + =
kPa , 1 P
B
34 038 =
Para la presin de roco (2.46) se tiene:
Condicin es
= 1 X
i
=
Sat
i i
P Y
1
P ; P = P
R
P
R
= 483,56 kPa
P
R
< P < P
B
Ya que la P del sistema est entre P
B
y P
R
, se procede a calcular L y V
40
Clculo de V por ecuacin (2.52):
41
( )
1 =
k z
i i
+ 1 k V 1
i
P
P
k
Sat
i
i
=
K
1
= 5,54
K
2
= 1,36
K
3
= 0,50
k
4
= 0,35
( )( ) ( )( ) ( )( ) ( )( )
1
65 0 1
35 0 05 0
5 0 1
50 0 60 0
36 0 1
36 1 20 0
54 4
54 5 1
=
+
+
+
+ V .
. .
V .
. .
V ,
, .
V , 1
, 5 0.
Se asumen valores de V hasta que se cumpla la igualdad.
V = 0.256 mol
Clculo de L:
L = 1 V = 1 0.744
L = 0.744 mol
Clculo de Y
i
, X
i
( ) 1 k V 1
k Z
y
i
i i
i
+
= y
i
i
i
k
Y
X =
021 , 0
345 , 0
249 , 0
384 , 0
4
3
2
1
=
=
=
=
y
y
y
y
0597 , 0
685 , 0
1836 , 0
07 , 0
4
3
2
1
=
=
=
=
x
x
x
x
Se cumple:
1 = =
i i
x y
42
Referencias
Balzhiser R. Michael R.S. Termodinmica Qumica para Ingenieros. Editorial
Pretince-Hall Internacional. 1974
PhD. Yolanda Reyes. Principios De Termodinmica Aplicada Para El rea De
Ingeniera Qumica.
Praunitz J.M. Molecular Thermodynamics of Fluids Phase Equilibria. Chap. 5,
Pretince-Hall. 1969.
Smith Van Nees. Introduccin a la Termodinmica en Ingeniera Qumica. 6ta
Edicin. 1996. Editorial McGraw Hill.
También podría gustarte
- Serena - Ron RashDocumento233 páginasSerena - Ron RashVioleta Matdamas Mendez0% (1)
- Salsa Tomate 080807Documento29 páginasSalsa Tomate 080807Violeta Matdamas MendezAún no hay calificaciones
- Nomograma de PriesterDocumento56 páginasNomograma de PriesterdanifexAún no hay calificaciones
- Respuesta dinámica de sistemas de primer y segundo ordenDocumento6 páginasRespuesta dinámica de sistemas de primer y segundo ordenVioleta Matdamas MendezAún no hay calificaciones
- SthephanopulousDocumento7 páginasSthephanopulousVioleta Matdamas MendezAún no hay calificaciones
- Forrest GumpDocumento138 páginasForrest GumpVioleta Matdamas Mendez80% (5)
- Proceso Tecnológico en La Producción de Conservas EsterilizadasDocumento13 páginasProceso Tecnológico en La Producción de Conservas EsterilizadasVioleta Matdamas MendezAún no hay calificaciones
- Salsa Tomate 080807Documento29 páginasSalsa Tomate 080807Violeta Matdamas MendezAún no hay calificaciones
- Nomograma de PriesterDocumento56 páginasNomograma de PriesterdanifexAún no hay calificaciones
- Ley de Nernst Marco TeóricoDocumento16 páginasLey de Nernst Marco TeóricoVioleta Matdamas MendezAún no hay calificaciones
- Antecedent EsDocumento2 páginasAntecedent EsVioleta Matdamas MendezAún no hay calificaciones
- 178 273 3 PBDocumento2 páginas178 273 3 PBVioleta Matdamas MendezAún no hay calificaciones
- 178 273 3 PBDocumento2 páginas178 273 3 PBVioleta Matdamas MendezAún no hay calificaciones
- Tallernac Adecuacionestructuras Ponencia2Documento14 páginasTallernac Adecuacionestructuras Ponencia2Violeta Matdamas MendezAún no hay calificaciones
- Produccion de Bioetanol A Partir de La Fermentación Alcoholica de Jarbes Glucosados Derivados de Cascaras de Naranja y Piña.Documento6 páginasProduccion de Bioetanol A Partir de La Fermentación Alcoholica de Jarbes Glucosados Derivados de Cascaras de Naranja y Piña.Ricardo Prieto Sandoval100% (1)
- Proteínas lácteas y sus propiedades funcionalesDocumento36 páginasProteínas lácteas y sus propiedades funcionalesVioleta Matdamas Mendez0% (1)
- RocioDocumento5 páginasRocioCami PereiraAún no hay calificaciones
- TFM Desire SantanaDocumento84 páginasTFM Desire Santanajes_sik778758Aún no hay calificaciones
- Cap7 Food - En.es PDFDocumento46 páginasCap7 Food - En.es PDFVioleta Matdamas MendezAún no hay calificaciones
- EnsayoDocumento2 páginasEnsayoVioleta Matdamas MendezAún no hay calificaciones
- Fluido SupercríticoDocumento7 páginasFluido SupercríticoVioleta Matdamas MendezAún no hay calificaciones
- Presión de VaporDocumento4 páginasPresión de VaporVioleta Matdamas MendezAún no hay calificaciones
- Ponchon SavaritDocumento26 páginasPonchon SavaritNahir Sarah Medina AntezanaAún no hay calificaciones
- The Prince PDFDocumento54 páginasThe Prince PDFLuna F. JonesAún no hay calificaciones
- File PDFDocumento1 páginaFile PDFVioleta Matdamas MendezAún no hay calificaciones
- Vapor LiquidoDocumento28 páginasVapor LiquidoDiego Esteban JarabaAún no hay calificaciones
- Problemas de PsicrometriaDocumento2 páginasProblemas de PsicrometriaDiomer CastroAún no hay calificaciones
- Cristalizacic 3 B 3 NDocumento21 páginasCristalizacic 3 B 3 NJuan Jose Arcce MaldonadoAún no hay calificaciones
- Guía Problemas Resueltos - Evaporadores Efecto Simple Versión Alfa1Documento26 páginasGuía Problemas Resueltos - Evaporadores Efecto Simple Versión Alfa1CuauRuvalkaba OrtizAún no hay calificaciones