También podría gustarte
- Tanque ImhoffDocumento16 páginasTanque Imhoffcr1525Aún no hay calificaciones
- Practica #6. Análisis Granulométricos Por Tamizado.Documento8 páginasPractica #6. Análisis Granulométricos Por Tamizado.Glendys MontielAún no hay calificaciones
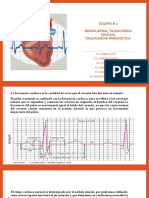
- Bradicardia Taquicardia Equipo #2Documento11 páginasBradicardia Taquicardia Equipo #2Alberto VazquezAún no hay calificaciones
- Ejemplo Cuadrado Latino Octubre 2020Documento6 páginasEjemplo Cuadrado Latino Octubre 2020delvis sanchez diasAún no hay calificaciones
- La Carne EnsayoDocumento4 páginasLa Carne EnsayoPedro Quiñones100% (1)
- Trucos SimsDocumento22 páginasTrucos SimsAlessia NottAún no hay calificaciones
- Identificacion de PCCDocumento5 páginasIdentificacion de PCCRuth Sofia Aguilar CcolqqueAún no hay calificaciones
- 8859Documento84 páginas8859Erik OñateAún no hay calificaciones
- Bases Administrativas para La Gestión de RiesgosDocumento20 páginasBases Administrativas para La Gestión de RiesgosANTONIO GARCIA HUERTAAún no hay calificaciones
- Circular Interna No. 6 Suspensión Del Contrato de TrabajoDocumento1 páginaCircular Interna No. 6 Suspensión Del Contrato de Trabajobrian natesAún no hay calificaciones
- Control de Publicidad de Medicamento1Documento12 páginasControl de Publicidad de Medicamento1sugeyAún no hay calificaciones
- Taller 3 UNRN Hidrocarburos Estructura PropiedadesDocumento6 páginasTaller 3 UNRN Hidrocarburos Estructura PropiedadesMore.e LopezAún no hay calificaciones
- Preservación y Conservación de Los Sistemas MecánicosDocumento9 páginasPreservación y Conservación de Los Sistemas MecánicosDioni ChavezAún no hay calificaciones
- Silabo - Fisiologia VgetalDocumento4 páginasSilabo - Fisiologia VgetalRodrich Guardia GAún no hay calificaciones
- CALCULO RENTA 4ta Categoria 2021Documento2 páginasCALCULO RENTA 4ta Categoria 2021MILAGROS YANE ACUÑA BRAVOAún no hay calificaciones
- Ataxia - AnatomiaDocumento12 páginasAtaxia - AnatomiaBelen GallardoAún no hay calificaciones
- 20 Metodos Analiticos para La Determinacion de Metabolitos Secundarios de PlantasDocumento108 páginas20 Metodos Analiticos para La Determinacion de Metabolitos Secundarios de PlantasAndres SernaAún no hay calificaciones
- UNFPA Guiaeducacionsexualintegral Unfpa Promsex F 1Documento97 páginasUNFPA Guiaeducacionsexualintegral Unfpa Promsex F 1sofiabloemAún no hay calificaciones
- Hongos, Algas y Virus (Ensayo)Documento5 páginasHongos, Algas y Virus (Ensayo)Araceli FabiánAún no hay calificaciones
- La Regresión Hipnótica Psicología o Fraude (Artículo) Autor Luis F. Díaz VilelaDocumento4 páginasLa Regresión Hipnótica Psicología o Fraude (Artículo) Autor Luis F. Díaz Vilelacarlos alfaro maciasAún no hay calificaciones
- Trastornos de Personalidad en Adictos A La CocainaDocumento49 páginasTrastornos de Personalidad en Adictos A La CocainaSol Solecito100% (3)
- Informe Económico Comité CentralDocumento2 páginasInforme Económico Comité CentralGerald Molina100% (1)
- Energias AlternativasDocumento2 páginasEnergias AlternativasMAXIMILIANO BALLEZA MARTINEZAún no hay calificaciones
- ANMAT Prohibió Comercialización de Limpiador MultiusoDocumento4 páginasANMAT Prohibió Comercialización de Limpiador MultiusoCrónicaAún no hay calificaciones
- Cuadro 1Documento1 páginaCuadro 1Oscar MestraAún no hay calificaciones
- Ficha Tecnica Celda SM6Documento2 páginasFicha Tecnica Celda SM6Yeison Steven HernanadezAún no hay calificaciones
- Subgrupo-2 Medicina LegalDocumento30 páginasSubgrupo-2 Medicina Legaladelaida muquincheAún no hay calificaciones
- Causes 2016Documento717 páginasCauses 2016Eli SalinasAún no hay calificaciones
- Reseña Histórica de La Herbolaria en MéxicoDocumento3 páginasReseña Histórica de La Herbolaria en MéxicoJazmin100% (2)
- Programa Oficial X Encuentro Estudiantil CneipDocumento8 páginasPrograma Oficial X Encuentro Estudiantil CneipRamón Velázquez RíosAún no hay calificaciones
- Recupera tu mente, reconquista tu vidaDe EverandRecupera tu mente, reconquista tu vidaCalificación: 5 de 5 estrellas5/5 (6)
- Yo Pude, ¡Tú Puedes!: Cómo tomar el control de tu bienestar emocional y convertirte en una persona imparable (edición revisada y expandida)De EverandYo Pude, ¡Tú Puedes!: Cómo tomar el control de tu bienestar emocional y convertirte en una persona imparable (edición revisada y expandida)Calificación: 5 de 5 estrellas5/5 (7)
- Cómo hacer que te pasen cosas buenas: Entiende tu cerebro, gestiona tus emociones, mejora tu vidaDe EverandCómo hacer que te pasen cosas buenas: Entiende tu cerebro, gestiona tus emociones, mejora tu vidaCalificación: 5 de 5 estrellas5/5 (1872)
- Psicología oscura: Una guía esencial de persuasión, manipulación, engaño, control mental, negociación, conducta humana, PNL y guerra psicológicaDe EverandPsicología oscura: Una guía esencial de persuasión, manipulación, engaño, control mental, negociación, conducta humana, PNL y guerra psicológicaCalificación: 4.5 de 5 estrellas4.5/5 (766)
- El Monje Que Vendio Su Ferrari: Una Fábula EspiritualDe EverandEl Monje Que Vendio Su Ferrari: Una Fábula EspiritualCalificación: 4.5 de 5 estrellas4.5/5 (1698)
- Tus Zonas Erroneas: Guía Para Combatir las Causas de la InfelicidadDe EverandTus Zonas Erroneas: Guía Para Combatir las Causas de la InfelicidadCalificación: 4.5 de 5 estrellas4.5/5 (1831)
- La revolución de la glucosa: Equilibra tus niveles de glucosa y cambiarás tu salud y tu vidaDe EverandLa revolución de la glucosa: Equilibra tus niveles de glucosa y cambiarás tu salud y tu vidaCalificación: 5 de 5 estrellas5/5 (203)
- Resetea tu mente. Descubre de lo que eres capazDe EverandResetea tu mente. Descubre de lo que eres capazCalificación: 5 de 5 estrellas5/5 (196)
- Los Secretos De La Mente Millonaria: Domina el juego de la riquezaDe EverandLos Secretos De La Mente Millonaria: Domina el juego de la riquezaCalificación: 5 de 5 estrellas5/5 (457)
- ¡Tómate un respiro! Mindfulness: El arte de mantener la calma en medio de la tempestadDe Everand¡Tómate un respiro! Mindfulness: El arte de mantener la calma en medio de la tempestadCalificación: 5 de 5 estrellas5/5 (198)