También podría gustarte
- Ensayo de EticaDocumento6 páginasEnsayo de EticaLaura StylesAún no hay calificaciones
- Informe 6. AntocianinasDocumento11 páginasInforme 6. AntocianinasLaura StylesAún no hay calificaciones
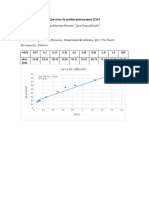
- Ejercicios de Análisis Instrumental 23114Documento3 páginasEjercicios de Análisis Instrumental 23114Laura Styles100% (1)
- EntropiaDocumento5 páginasEntropiaLaura Styles100% (1)
- Apuntes de InmunologiaDocumento3 páginasApuntes de InmunologiaLaura StylesAún no hay calificaciones
- Guia 7 Determinación Del Punto Isolectrico y La Capacidad Buffer de Un Amino AcidoDocumento2 páginasGuia 7 Determinación Del Punto Isolectrico y La Capacidad Buffer de Un Amino AcidoLaura StylesAún no hay calificaciones
- Taller PDF 11Documento22 páginasTaller PDF 11Laura StylesAún no hay calificaciones
- Taller de Coloración o Tincion de GramDocumento1 páginaTaller de Coloración o Tincion de GramLaura StylesAún no hay calificaciones
- Preparacion de SolucionesDocumento5 páginasPreparacion de SolucionesLaura StylesAún no hay calificaciones
- EstandarizacionDocumento3 páginasEstandarizacionLaura StylesAún no hay calificaciones
- Calor de ReaccionDocumento6 páginasCalor de ReaccionLaura StylesAún no hay calificaciones
- Preparacion de SolucionesDocumento5 páginasPreparacion de SolucionesLaura StylesAún no hay calificaciones
- Calibracion Del CalorimetroDocumento7 páginasCalibracion Del CalorimetroLaura StylesAún no hay calificaciones
- Leyes de Los GasesDocumento8 páginasLeyes de Los GasesLaura StylesAún no hay calificaciones
- Sistemas de Captación en Caudales BajosDocumento10 páginasSistemas de Captación en Caudales BajosManuelsoAún no hay calificaciones
- Guia Microanato 6Documento6 páginasGuia Microanato 6gvmajano.04Aún no hay calificaciones
- RVCDocumento7 páginasRVCapi-394126947Aún no hay calificaciones
- Trabajo de Fisica PolDocumento4 páginasTrabajo de Fisica PolJuan Manuel CavaAún no hay calificaciones
- Soy Adolescente Entiendeme Angela Marulanda GomezDocumento37 páginasSoy Adolescente Entiendeme Angela Marulanda GomezPaula Monroy100% (2)
- Sensor Pir ArduinoDocumento4 páginasSensor Pir ArduinojbuabudAún no hay calificaciones
- Nazca y Su Cultura para Sexto de PrimariaDocumento3 páginasNazca y Su Cultura para Sexto de PrimariaAna Ysabel PeredaAún no hay calificaciones
- 01 Sistema de Medición Angular Parte 2Documento1 página01 Sistema de Medición Angular Parte 2Wilber Ramos CartAún no hay calificaciones
- Final - Programa Nacional de Salud de La Infancia Con Enfoque IntegralDocumento12 páginasFinal - Programa Nacional de Salud de La Infancia Con Enfoque IntegralMatias ArenasAún no hay calificaciones
- Tareas para El Desarrollo de Habilidades de Visualización y Orientación EspacialDocumento19 páginasTareas para El Desarrollo de Habilidades de Visualización y Orientación EspacialClauconejerosAún no hay calificaciones
- Cochrane - Specificacioìn (General) ConsultasDocumento1 páginaCochrane - Specificacioìn (General) ConsultasJorge Antonio Díaz NambrardAún no hay calificaciones
- Factura Fecv - 929 Comercialixadora Macoser de Occidente SasDocumento1 páginaFactura Fecv - 929 Comercialixadora Macoser de Occidente Sasluis miguel rios taveraAún no hay calificaciones
- Mgia Visual - Jan Fries PasDocumento64 páginasMgia Visual - Jan Fries Paskaliyuga123Aún no hay calificaciones
- Procesos Y Procedimientos LogísticosDocumento15 páginasProcesos Y Procedimientos Logísticosmarketer201067% (6)
- Ejercicios Pruebas de HipotesisDocumento9 páginasEjercicios Pruebas de HipotesisSergio MoreiraAún no hay calificaciones
- Historia de La Fotografía PublicitariaDocumento39 páginasHistoria de La Fotografía PublicitariaKaren BARRETO ÁLVAREZ100% (1)
- Land ArtDocumento19 páginasLand ArtsandraxsxvAún no hay calificaciones
- Unidad 4Documento31 páginasUnidad 4JHONATANAún no hay calificaciones
- Características Macroscópicas y Microscópicas en Un HongoDocumento23 páginasCaracterísticas Macroscópicas y Microscópicas en Un HongoSamya Polet Meléndez Cruz43% (7)
- 2 SomatotipoDocumento10 páginas2 SomatotipoCinthyaLissethAlejandroAún no hay calificaciones
- C02_-_Manual_Tea_Sommelier_impDocumento53 páginasC02_-_Manual_Tea_Sommelier_impCeleste G.Aún no hay calificaciones
- Pip MTC 2234980Documento269 páginasPip MTC 2234980thagher100% (1)
- Resumen Ocampo, Arzeno y GrassanoDocumento10 páginasResumen Ocampo, Arzeno y GrassanoSol VendittiAún no hay calificaciones
- Extracción de Colorante de AchioteDocumento45 páginasExtracción de Colorante de AchioteMarita M. Orbegoso100% (2)
- Tarea 7 Electro. I 2020 B ISE y Titulac PotenciomDocumento3 páginasTarea 7 Electro. I 2020 B ISE y Titulac PotenciomAlex CisnerosAún no hay calificaciones
- Asi Se Dice Level 1 - WorkbookDocumento263 páginasAsi Se Dice Level 1 - Workbookzephry50% (4)
- Sesion de Aprendizaje 3 - Inyectoterapia - Carga de MedDocumento38 páginasSesion de Aprendizaje 3 - Inyectoterapia - Carga de MedDanitza MelendrezAún no hay calificaciones
- Finanzas TareaDocumento20 páginasFinanzas TareaCarlos VargasAún no hay calificaciones
- Captura de Pantalla 2021-12-08 A La(s) 12.53.14Documento28 páginasCaptura de Pantalla 2021-12-08 A La(s) 12.53.14Rodrigo Pujol Del ToroAún no hay calificaciones
- Guia #9Documento9 páginasGuia #9EmersonAún no hay calificaciones