También podría gustarte
- Guia 1 EndocrinologiaDocumento3 páginasGuia 1 EndocrinologiaAlezie Roosevelt Paul GarciaAún no hay calificaciones
- Informe BCT II - Músculo LisoDocumento14 páginasInforme BCT II - Músculo Lisodiebalo1704Aún no hay calificaciones
- Atresia AnalDocumento51 páginasAtresia AnalJosue CamposAún no hay calificaciones
- Nervios A Los Que Anestesiamos en Odontologia.Documento5 páginasNervios A Los Que Anestesiamos en Odontologia.Maritza. Rios vásquezAún no hay calificaciones
- Taller de Septimo de Agropecuaria PlantasDocumento2 páginasTaller de Septimo de Agropecuaria PlantasRoger Stevenson Ruiz SeguraAún no hay calificaciones
- Tema: La SangreDocumento5 páginasTema: La SangreM.Jesús Zapana VargasAún no hay calificaciones
- Nerv IosDocumento1 páginaNerv IosDaniel GonzalezAún no hay calificaciones
- Factores Reguladores de La Resorción ÓseaDocumento16 páginasFactores Reguladores de La Resorción ÓseaADRIANA PASTRANAAún no hay calificaciones
- Sistema Tegumentario: La Piel y sus FuncionesDocumento15 páginasSistema Tegumentario: La Piel y sus FuncionesMelba NoboaAún no hay calificaciones
- Glosario Del Sistema RespiratorioDocumento7 páginasGlosario Del Sistema RespiratorioNoelia Alfonsina Montero Vaca100% (1)
- Espermatobioscopia: examen de fertilidad masculinaDocumento6 páginasEspermatobioscopia: examen de fertilidad masculinaNeftalíAún no hay calificaciones
- FagocitosisDocumento3 páginasFagocitosisMariana OsorioAún no hay calificaciones
- Folleto de Lesiones Más Comunes en El Trabajo o Vida Cotidiana, y La Importancia de Las Pausas Activas GA6-230101507-AA4-EV01Documento3 páginasFolleto de Lesiones Más Comunes en El Trabajo o Vida Cotidiana, y La Importancia de Las Pausas Activas GA6-230101507-AA4-EV01Angie SanchezAún no hay calificaciones
- Des OseoDocumento13 páginasDes OseoJazmin AlvarezAún no hay calificaciones
- Locomocion en Los Seres VivosDocumento1 páginaLocomocion en Los Seres VivosILDIFONSO ROCA ROMAN100% (1)
- LIPIDOSDocumento27 páginasLIPIDOSJOEL ANDRE SANCHEZ GILAún no hay calificaciones
- Programa y Guía de T. Particos - Citología e Histología VegetalDocumento37 páginasPrograma y Guía de T. Particos - Citología e Histología VegetalJaqueline ToledoAún no hay calificaciones
- Proceso fisiológico del puerperio bovinoDocumento22 páginasProceso fisiológico del puerperio bovinoAlan RojasAún no hay calificaciones
- Org ExpoDocumento9 páginasOrg ExpoDANIELA ALEJANDRA ORTIZ ALVAREZAún no hay calificaciones
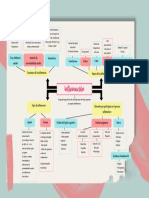
- Mapa Conceptual InflamaciónDocumento1 páginaMapa Conceptual InflamaciónMaría Fernanda Argote100% (6)
- Mapa Conceptual de La Celula 6Documento1 páginaMapa Conceptual de La Celula 6vc8jhp65vr100% (1)
- Sistema DigestivoDocumento34 páginasSistema DigestivoANGHELA JHANINA MONCADA VALLEJOSAún no hay calificaciones
- Respuestas y Adaptaciones en El Ejercicio Guido SicilianiDocumento14 páginasRespuestas y Adaptaciones en El Ejercicio Guido SicilianiDANIEL ALEJANDRO100% (1)
- Cuáles Son Las Subramas de La FísicaDocumento3 páginasCuáles Son Las Subramas de La FísicaOswaldo Gámez100% (1)
- Manual Usuario Bc-20s HematologiaDocumento173 páginasManual Usuario Bc-20s HematologiaNARIÑO CONSACAAún no hay calificaciones
- Articulaciones Del Tronco y TóraxDocumento39 páginasArticulaciones Del Tronco y Tórax929400428cocoAún no hay calificaciones
- Ficha FinalDocumento2 páginasFicha FinalJessy RodriguezAún no hay calificaciones
- Taller T7 Efectos de CalorDocumento5 páginasTaller T7 Efectos de CalorStiven VasquezAún no hay calificaciones
- Práctica Estructuras VegetalesDocumento22 páginasPráctica Estructuras VegetalesFernando MosqueraAún no hay calificaciones
- Hábitos Que Afectan Mi Entorno Personal y AcadémicoDocumento1 páginaHábitos Que Afectan Mi Entorno Personal y AcadémicoDANNA NICOLLE SAUCEDO SANCHEZAún no hay calificaciones