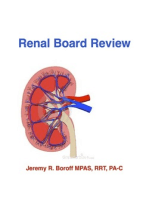
También podría gustarte
- ACP Board Review Nephrology 20052ndDocumento35 páginasACP Board Review Nephrology 20052nddoctormido2010100% (1)
- Statistics & Molecular MRCP1Documento87 páginasStatistics & Molecular MRCP1Raouf Ra'fat Soliman100% (2)
- Busyspr MRCPDocumento38 páginasBusyspr MRCPNasir AyubAún no hay calificaciones
- Rheumatology MRCP1Documento186 páginasRheumatology MRCP1Raouf Ra'fat Soliman100% (3)
- MRCP Exam TipsDocumento7 páginasMRCP Exam Tipsshaaish100% (2)
- MRCP Revision NotesDocumento14 páginasMRCP Revision NotesSharanyMustafaAún no hay calificaciones
- UntitledDocumento219 páginasUntitledSagit Nauman81Aún no hay calificaciones
- Murray CaseStudiesInNephrologyDocumento179 páginasMurray CaseStudiesInNephrologyAdil BilaeloAún no hay calificaciones
- Magdy AbbasDocumento49 páginasMagdy AbbasAli AlisonAún no hay calificaciones
- Nephrology Curriculum - الزمالة المصرية للكليDocumento60 páginasNephrology Curriculum - الزمالة المصرية للكليbook1man100% (1)
- FRACP Recall Paper 2002: Part A - 70 Questions, 2 HoursDocumento43 páginasFRACP Recall Paper 2002: Part A - 70 Questions, 2 HoursZH. omg sarAún no hay calificaciones
- MRCPass Notes For MRCP 1 - HEMATOLOGYDocumento9 páginasMRCPass Notes For MRCP 1 - HEMATOLOGYsabdali100% (1)
- HematoboardreviewDocumento45 páginasHematoboardreviewRapid MedicineAún no hay calificaciones
- The Ultimate Revision Basic Science: BY DR - Sherif Badrawy Critical CareDocumento685 páginasThe Ultimate Revision Basic Science: BY DR - Sherif Badrawy Critical CareSherif Elbadrawy100% (1)
- Pituitary and Endocrine Gland Disorders NotesDocumento12 páginasPituitary and Endocrine Gland Disorders Notessabdali100% (1)
- SABER IM PrometricDocumento71 páginasSABER IM Prometricisra zaidAún no hay calificaciones
- Nephrology PDFDocumento38 páginasNephrology PDFOmar AyashAún no hay calificaciones
- 17 - Toronto Notes 2011 - NephrologyDocumento92 páginas17 - Toronto Notes 2011 - NephrologyKhairulsani Yusof100% (2)
- Step 3 Nephrology Part 1 of 3Documento7 páginasStep 3 Nephrology Part 1 of 3Bhavin ChoksiAún no hay calificaciones
- Badrawy MRCP RevisionDocumento14 páginasBadrawy MRCP RevisionSharanyMustafaAún no hay calificaciones
- Lateral thinking in MRCP PACES examDocumento73 páginasLateral thinking in MRCP PACES examSagit Nauman81100% (1)
- NEPHROURO EXAMDocumento22 páginasNEPHROURO EXAMRishi KNair100% (1)
- MRCP Paces Chest RedclifffDocumento13 páginasMRCP Paces Chest RedclifffIsmail H AAún no hay calificaciones
- EMQs and MCQs For Medical Finals (PDFDrive)Documento337 páginasEMQs and MCQs For Medical Finals (PDFDrive)Samuel TanAún no hay calificaciones
- Toronto Notes Nephrology 2015 35Documento1 páginaToronto Notes Nephrology 2015 35JUSASB50% (2)
- Acp Sglt2 Slidecast 220Documento169 páginasAcp Sglt2 Slidecast 220charanmann9165Aún no hay calificaciones
- Manual of Clinical Nephrology by Rafiqul AlamDocumento12 páginasManual of Clinical Nephrology by Rafiqul AlamSELLULARAún no hay calificaciones
- Optimising Success in FRCEM Intermediate SAQsDocumento109 páginasOptimising Success in FRCEM Intermediate SAQsSricharan SairiAún no hay calificaciones
- Emedica MRCGP AKT Curriculum ChecklistDocumento42 páginasEmedica MRCGP AKT Curriculum ChecklistSana Mustafa100% (1)
- 5 Nephrology MRCP 1 2019 Q BankDocumento931 páginas5 Nephrology MRCP 1 2019 Q BankRuby BhattyAún no hay calificaciones
- Rash BookDocumento12 páginasRash BookPhoebe UsmleAún no hay calificaciones
- Internal Medicine 1 Conrad FischerDocumento35 páginasInternal Medicine 1 Conrad Fischerjaber fathiAún no hay calificaciones
- Prof Emma-Edit Endocrine Hypertension Meet The ExpertsDocumento32 páginasProf Emma-Edit Endocrine Hypertension Meet The ExpertsJames KomalingAún no hay calificaciones
- Mock Test-Free For MRCP (Uk) - 1 by Sumanta Sir - S MRCPDocumento10 páginasMock Test-Free For MRCP (Uk) - 1 by Sumanta Sir - S MRCPxuexueAún no hay calificaciones
- Coronary Artery DiseaseDocumento3 páginasCoronary Artery DiseaseRitz CelsoAún no hay calificaciones
- Endocrine, Metabolic & Nephrology MCQS: Thyroid Eye DiseaseDocumento78 páginasEndocrine, Metabolic & Nephrology MCQS: Thyroid Eye DiseaseDr.younes95 RekaaneyAún no hay calificaciones
- January 2014 MRCP 1Documento201 páginasJanuary 2014 MRCP 1Sherif ElbadrawyAún no hay calificaciones
- Nephrology ExamsDocumento30 páginasNephrology Examsdhianne_garcia2001100% (1)
- CALLED.TO.SEE.PATIENT v1.1 survival guideDocumento111 páginasCALLED.TO.SEE.PATIENT v1.1 survival guideGlen OngAún no hay calificaciones
- Nephrology Certification Examination Blueprint - American Board of Internal MedicineDocumento4 páginasNephrology Certification Examination Blueprint - American Board of Internal MedicineabimorgAún no hay calificaciones
- MRCP 1 Common-Topics-Sep-2019 PDFDocumento1 páginaMRCP 1 Common-Topics-Sep-2019 PDFZubair Mahmood KamalAún no hay calificaciones
- EMERGENCY MEDICINE IN-SERVICE EXAM REVIEW 2009Documento147 páginasEMERGENCY MEDICINE IN-SERVICE EXAM REVIEW 2009iamo107501100% (1)
- MRCP Part 1Documento1 páginaMRCP Part 1Ali AlisonAún no hay calificaciones
- MRCP 1 Last MinDocumento10 páginasMRCP 1 Last MinDr.yeasin Arafat100% (1)
- MRCP Part 1-Pharm GuestionsDocumento42 páginasMRCP Part 1-Pharm GuestionswyenyAún no hay calificaciones
- Diagnosing Gastrointestinal and Seizure Disorders from Clinical VignettesDocumento33 páginasDiagnosing Gastrointestinal and Seizure Disorders from Clinical VignettesSean Purrier50% (2)
- Assem Draz: Neurological DisordersDocumento26 páginasAssem Draz: Neurological DisordersxuexueAún no hay calificaciones
- Core Topics in Internal MedicineDocumento4 páginasCore Topics in Internal MedicineKristina Anne CoAún no hay calificaciones
- Beyond Basic NephrologyDocumento1 páginaBeyond Basic NephrologyexpensivecareAún no hay calificaciones
- Nekc Lump PacesDocumento5 páginasNekc Lump PacesTahir AliAún no hay calificaciones
- Recall MRCP 1 Jan 2014Documento20 páginasRecall MRCP 1 Jan 2014Mohamed Amr SalamaAún no hay calificaciones
- IV PDFDocumento63 páginasIV PDFelbagouryAún no hay calificaciones
- Cardiac TestsDocumento6 páginasCardiac TestsRobert DowneyAún no hay calificaciones
- Behav SC Board RevDocumento55 páginasBehav SC Board RevyaykataiAún no hay calificaciones
- Environmental, Congenital, and Iatrogenic Syndrome GuideDocumento4 páginasEnvironmental, Congenital, and Iatrogenic Syndrome GuideFranspolAún no hay calificaciones
- Git 2 Part 1 (Manifestations of Liver Cell Failure) UploadDocumento66 páginasGit 2 Part 1 (Manifestations of Liver Cell Failure) UploadRaouf Ra'fat SolimanAún no hay calificaciones
- Neuro TestDocumento9 páginasNeuro TestHanung MerahbaraAún no hay calificaciones
- Cranial Nerves Anatomy Review NoteDocumento5 páginasCranial Nerves Anatomy Review NoteOlu FasanyaAún no hay calificaciones
- Antibiotic Guidelines 2012Documento100 páginasAntibiotic Guidelines 2012HelenAún no hay calificaciones
- Pharmacology SummaryDocumento16 páginasPharmacology Summarysechzhen96% (46)
- Tutti FruttiDocumento10 páginasTutti FruttishadapaaakAún no hay calificaciones
- Reference ID: 3418715: Sections or Subsections Omitted From The Full Prescribing Information Are Not ListedDocumento34 páginasReference ID: 3418715: Sections or Subsections Omitted From The Full Prescribing Information Are Not ListedRaouf Ra'fat SolimanAún no hay calificaciones
- GIT 3 Part 1 (HepatitisDocumento51 páginasGIT 3 Part 1 (HepatitisRaouf Ra'fat SolimanAún no hay calificaciones
- JaundiceDocumento5 páginasJaundiceRaouf Ra'fat Soliman100% (1)
- Git 2 Part 2 (Hepatitis) Upload PDFDocumento30 páginasGit 2 Part 2 (Hepatitis) Upload PDFRaouf Ra'fat SolimanAún no hay calificaciones
- GIT 3 Part 2 (Cirrhosis - Chronic Hepatitis - G.I.T.) Upload PDFDocumento76 páginasGIT 3 Part 2 (Cirrhosis - Chronic Hepatitis - G.I.T.) Upload PDFRaouf Ra'fat SolimanAún no hay calificaciones
- EndocrineDocumento181 páginasEndocrineRaouf Ra'fat SolimanAún no hay calificaciones
- GIT4 - PART2 (Malabsorption Syndrome - Jaundice) Upload PDFDocumento60 páginasGIT4 - PART2 (Malabsorption Syndrome - Jaundice) Upload PDFRaouf Ra'fat SolimanAún no hay calificaciones
- GIT 4 - PART1 (IBD - Ascites - Functional Gastrointestinal Disorders) Upload PDFDocumento45 páginasGIT 4 - PART1 (IBD - Ascites - Functional Gastrointestinal Disorders) Upload PDFRaouf Ra'fat SolimanAún no hay calificaciones
- Chest - DR Abo-ElAsrar - by El Azhar Medical Students 2012Documento56 páginasChest - DR Abo-ElAsrar - by El Azhar Medical Students 2012Fatma HelalAún no hay calificaciones
- GIT4 - PART3 (Cases) Upload PDFDocumento9 páginasGIT4 - PART3 (Cases) Upload PDFRaouf Ra'fat SolimanAún no hay calificaciones
- Cardiology Scheme and Heart FailureDocumento76 páginasCardiology Scheme and Heart FailureRaouf Ra'fat SolimanAún no hay calificaciones
- Git 1 (Liver Function) Upload PDFDocumento37 páginasGit 1 (Liver Function) Upload PDFRaouf Ra'fat SolimanAún no hay calificaciones
- Acid Base Balance DR Ahmad Mowafy Revision UpdatedDocumento18 páginasAcid Base Balance DR Ahmad Mowafy Revision UpdatedRaouf Ra'fat SolimanAún no hay calificaciones
- Genetics - DR Abo-ElAsrar - by El Azhar Medical Students 2012Documento39 páginasGenetics - DR Abo-ElAsrar - by El Azhar Medical Students 2012Raouf Ra'fat Soliman100% (1)
- InfectionDocumento294 páginasInfectionRaouf Ra'fat SolimanAún no hay calificaciones
- Asrar - Ped - Clinical - DMP3Documento73 páginasAsrar - Ped - Clinical - DMP3Raouf Ra'fat Soliman100% (1)
- Crdiology - DR Abo-ElAsrar - by El Azhar Medical Students 2012Documento74 páginasCrdiology - DR Abo-ElAsrar - by El Azhar Medical Students 2012Raouf Ra'fat SolimanAún no hay calificaciones
- Infection Control in ArabicDocumento105 páginasInfection Control in ArabicRaouf Ra'fat SolimanAún no hay calificaciones
- NutritionDocumento354 páginasNutritionRaouf Ra'fat SolimanAún no hay calificaciones
- Rheumatology MRCP1Documento186 páginasRheumatology MRCP1Raouf Ra'fat Soliman100% (3)
- Analyte and assay sheet regulatory affairsDocumento850 páginasAnalyte and assay sheet regulatory affairsAbid Ali KhanAún no hay calificaciones
- Patients' Expectation and Satisfaction With Complete Denture Before and After The TherapyDocumento4 páginasPatients' Expectation and Satisfaction With Complete Denture Before and After The TherapyRespiika YuliaaAún no hay calificaciones
- Precision Attachments For The 21st CenturyDocumento6 páginasPrecision Attachments For The 21st CenturyMohsin Habib67% (3)
- Cardio, Otis, Ortho, and GI Drug ListsDocumento4 páginasCardio, Otis, Ortho, and GI Drug ListsJodi Gugel DeMarrowAún no hay calificaciones
- Question of The Week # 433: Archer Red 11:25 AM Links To This PostDocumento34 páginasQuestion of The Week # 433: Archer Red 11:25 AM Links To This PostChristian JaraAún no hay calificaciones
- Headache PDFDocumento92 páginasHeadache PDFRohamonangan TheresiaAún no hay calificaciones
- Managing The MismatchDocumento36 páginasManaging The MismatchhumansamirAún no hay calificaciones
- 1111111Documento9 páginas1111111kooli koolantAún no hay calificaciones
- Pedia Nr-4 2013 Art-8Documento10 páginasPedia Nr-4 2013 Art-8Daniela CioboataAún no hay calificaciones
- Medicina de Urgencias Pediatricas de Strange 1era EdicionDocumento810 páginasMedicina de Urgencias Pediatricas de Strange 1era EdicionGerardo Acosta Galvan100% (1)
- High Yield Surgery Shelf Exam Review CompleteDocumento10 páginasHigh Yield Surgery Shelf Exam Review CompleteAmir Ali100% (1)
- Unit 236 MedicationDocumento2 páginasUnit 236 MedicationSzabolcs Lehota60% (5)
- Psychiatric Interview (Book Review) PDFDocumento274 páginasPsychiatric Interview (Book Review) PDFihsansabridr100% (3)
- Incident Reporting - NSL3.1 (Apr 2011)Documento16 páginasIncident Reporting - NSL3.1 (Apr 2011)fwisheeAún no hay calificaciones
- DIR Spring 2016 ONLINE Boards Fodder Lab WorkupsDocumento4 páginasDIR Spring 2016 ONLINE Boards Fodder Lab WorkupsJamesAún no hay calificaciones
- Sedation Concept Map 2 PDFDocumento3 páginasSedation Concept Map 2 PDFAlvin L. RozierAún no hay calificaciones
- LTCW Breast Write UpDocumento21 páginasLTCW Breast Write Upapi-632682404Aún no hay calificaciones
- Diificult AirwayDocumento13 páginasDiificult AirwayBhi-An BatobalonosAún no hay calificaciones
- SLE1000 NCPAP Generator BrochureDocumento4 páginasSLE1000 NCPAP Generator BrochureLASINRANGAún no hay calificaciones
- 3rd Year Precept Pedia Case 4 HydroceleDocumento4 páginas3rd Year Precept Pedia Case 4 Hydrocelekristel_nicole18yaho100% (1)
- Jawrelationsincompletedentures+ Audio 130425145804 Phpapp01Documento42 páginasJawrelationsincompletedentures+ Audio 130425145804 Phpapp01Islam ul haqAún no hay calificaciones
- Hospital Information SystemDocumento2 páginasHospital Information SystemManpreetaaAún no hay calificaciones
- State by State Nurse Practitioner RequirementsDocumento29 páginasState by State Nurse Practitioner RequirementsRick Whitley100% (1)
- Vitamin K Shot Prevents Bleeds in BabiesDocumento11 páginasVitamin K Shot Prevents Bleeds in BabiesLucky PuspitasariAún no hay calificaciones
- BMUS Safety Guidelines 2009 Revision FINAL Nov 2009 PDFDocumento18 páginasBMUS Safety Guidelines 2009 Revision FINAL Nov 2009 PDFeryxspAún no hay calificaciones
- ACC AHA ACE 2003 Guideline Update For The Clinical PDFDocumento99 páginasACC AHA ACE 2003 Guideline Update For The Clinical PDFsaveclipAún no hay calificaciones
- Untitled PDFDocumento8 páginasUntitled PDFAndrei BriceagAún no hay calificaciones
- Boala Paget A Glandei MamareDocumento41 páginasBoala Paget A Glandei MamarePANNADYAún no hay calificaciones
- Medicall Private Practice Final PDFDocumento109 páginasMedicall Private Practice Final PDFRohit BharadwajAún no hay calificaciones
- Reaction PaperDocumento3 páginasReaction Paperedmond callenAún no hay calificaciones