También podría gustarte
- Caso ClinicoDocumento8 páginasCaso ClinicoDayanaOlgaTorresHuamaniAún no hay calificaciones
- Articulo de BioseguridadDocumento41 páginasArticulo de BioseguridadDayanaOlgaTorresHuamaniAún no hay calificaciones
- Cardiovascular SystemDocumento6 páginasCardiovascular SystemDayanaOlgaTorresHuamaniAún no hay calificaciones
- Caso Clinico 12-12-22 Cardiologia DR RevattaDocumento4 páginasCaso Clinico 12-12-22 Cardiologia DR RevattaDayanaOlgaTorresHuamaniAún no hay calificaciones
- Intoxicacion Por OrganofosforadosDocumento3 páginasIntoxicacion Por OrganofosforadosDayanaOlgaTorresHuamaniAún no hay calificaciones
- Conceptos Básicos de Patología SEMINARIO GRUPO DDocumento10 páginasConceptos Básicos de Patología SEMINARIO GRUPO DDayanaOlgaTorresHuamaniAún no hay calificaciones
- 20 Al 28 en EspañolDocumento3 páginas20 Al 28 en EspañolDayanaOlgaTorresHuamaniAún no hay calificaciones
- Amiloidosis y Gota SeminarioDocumento16 páginasAmiloidosis y Gota SeminarioDayanaOlgaTorresHuamaniAún no hay calificaciones
- ICTERICIADocumento2 páginasICTERICIADayanaOlgaTorresHuamaniAún no hay calificaciones
- Inducción e Inhibición Enzimática de MedicamentosDocumento1 páginaInducción e Inhibición Enzimática de MedicamentosDayanaOlgaTorresHuamaniAún no hay calificaciones
- CUESTIONARIO DE SÍNTOMAS SRQ-psicopedagogiaDocumento2 páginasCUESTIONARIO DE SÍNTOMAS SRQ-psicopedagogiaDayanaOlgaTorresHuamaniAún no hay calificaciones
- 1 Definición y Etiología de IctericiaDocumento2 páginas1 Definición y Etiología de IctericiaDayanaOlgaTorresHuamaniAún no hay calificaciones
- Esquema de VacunaciónDocumento12 páginasEsquema de VacunaciónDayanaOlgaTorresHuamaniAún no hay calificaciones
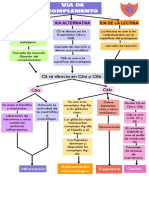
- Via de ComplementoDocumento1 páginaVia de ComplementoDayanaOlgaTorresHuamaniAún no hay calificaciones
- Conceptos Básicos de Patología SEMINARIO GRUPO DDocumento10 páginasConceptos Básicos de Patología SEMINARIO GRUPO DDayanaOlgaTorresHuamaniAún no hay calificaciones
- Variabilidad Biológica y Efectos Indeseables de Los FármacosDocumento3 páginasVariabilidad Biológica y Efectos Indeseables de Los FármacosDayanaOlgaTorresHuamaniAún no hay calificaciones
- 1 Definición y Etiología de IctericiaDocumento2 páginas1 Definición y Etiología de IctericiaDayanaOlgaTorresHuamaniAún no hay calificaciones
- Ecografia Normal de HombroDocumento8 páginasEcografia Normal de HombronatycaflAún no hay calificaciones
- Ricardo Koenig PDFDocumento43 páginasRicardo Koenig PDFjavi222222Aún no hay calificaciones
- Aspiracion de Secreciones Abierta y CerradaDocumento12 páginasAspiracion de Secreciones Abierta y CerradaCarlos J Marin NaalAún no hay calificaciones
- Prevención Psicológica ComunitariaDocumento10 páginasPrevención Psicológica ComunitariaDana SasschettiAún no hay calificaciones
- Grupo 1Documento14 páginasGrupo 1DANIELAún no hay calificaciones
- 526.sexo y Juventud - Pérez SorianoDocumento131 páginas526.sexo y Juventud - Pérez SorianoleydiAún no hay calificaciones
- Dolor Tbe 3pDocumento3 páginasDolor Tbe 3pEneko LandaburuAún no hay calificaciones
- Cuadro de Control Insumos IchuDocumento62 páginasCuadro de Control Insumos Ichuchristian1909Aún no hay calificaciones
- Sesiã"n 1 Ev. Psicopedagã"Gica.Documento3 páginasSesiã"n 1 Ev. Psicopedagã"Gica.Diego CantoralAún no hay calificaciones
- Farmacocinetica 2 4327Documento42 páginasFarmacocinetica 2 4327Luis LuisinAún no hay calificaciones
- Preparación de Medio de Cultivo y SiembraDocumento15 páginasPreparación de Medio de Cultivo y SiembraAnthonyAún no hay calificaciones
- CAP3.-El-deterioro-cognitivo-y-las-demencias. NuevoDocumento10 páginasCAP3.-El-deterioro-cognitivo-y-las-demencias. NuevoManu CavalleraAún no hay calificaciones
- Extracción de La Bola Adiposa de BichatDocumento2 páginasExtracción de La Bola Adiposa de BichatNicolasAún no hay calificaciones
- Anemia megaloblastica por déficit de vitamina B12 y ácido fólicoDocumento19 páginasAnemia megaloblastica por déficit de vitamina B12 y ácido fólicoYesenia Aide Vivar AlvaAún no hay calificaciones
- Problemáticas Seleccionadas Sobre A.T. en Pacientes Adictos. T.P ELENADocumento10 páginasProblemáticas Seleccionadas Sobre A.T. en Pacientes Adictos. T.P ELENAMaria Marcela CarpentierAún no hay calificaciones
- Fortalecimiento de la salud músculoesquelética en el trabajoDocumento1 páginaFortalecimiento de la salud músculoesquelética en el trabajoDairo RuedaAún no hay calificaciones
- Salud Ocupacional: Universidad Nacional Santiago Del Estero Facultad de Ciencias Médicas AsignaturaDocumento18 páginasSalud Ocupacional: Universidad Nacional Santiago Del Estero Facultad de Ciencias Médicas AsignaturaMily Lopez GomezAún no hay calificaciones
- Pma OxapampaDocumento46 páginasPma OxapampajessicaAún no hay calificaciones
- Fármacos Antiadrenérgicos - Efectos Sistémicos, Usos TerapéuticosDocumento28 páginasFármacos Antiadrenérgicos - Efectos Sistémicos, Usos Terapéuticosmauro soudrecamposAún no hay calificaciones
- Úlceras Por PresiónDocumento8 páginasÚlceras Por Presiónsusana andradeAún no hay calificaciones
- Fisiologia Vegetal IIIDocumento70 páginasFisiologia Vegetal IIIAlexandra AlvarezAún no hay calificaciones
- Proyecto Individual San AntonDocumento17 páginasProyecto Individual San AntonJESUS PEREZ AGUILARAún no hay calificaciones
- Trabajo de Disertacion Impacto AmbientalDocumento36 páginasTrabajo de Disertacion Impacto AmbientalJimmy MoraAún no hay calificaciones
- EXT wcUvywrGuMrDUULxWrkP PDFDocumento6 páginasEXT wcUvywrGuMrDUULxWrkP PDFDani M0% (1)
- Marco Teorico y Antecedentes-EthelDocumento22 páginasMarco Teorico y Antecedentes-EtheljeancAún no hay calificaciones
- Resumen Del Artículo Katalin KarikóDocumento2 páginasResumen Del Artículo Katalin KarikóFREDDY JOHAN SILVA RUIZAún no hay calificaciones
- Actividad 1 Pieza ComunicativaDocumento10 páginasActividad 1 Pieza ComunicativaAngel AndreyAún no hay calificaciones
- Trabajo Practico, Manipulacion de Especialidades FarmaceuticasDocumento10 páginasTrabajo Practico, Manipulacion de Especialidades FarmaceuticasMARIA ELENA JARA RENGIFOAún no hay calificaciones
- Tratamiento Ablativo Basado en La Técnica de TermoDocumento46 páginasTratamiento Ablativo Basado en La Técnica de TermoJostin Power100% (1)
- Manual de Acupuntura y Digitopuntura para El Médic - 221120 - 225417 PDFDocumento106 páginasManual de Acupuntura y Digitopuntura para El Médic - 221120 - 225417 PDFsaraiAún no hay calificaciones
- Ansiedad infantil. Los trastornos explicados a los padresDe EverandAnsiedad infantil. Los trastornos explicados a los padresCalificación: 4.5 de 5 estrellas4.5/5 (25)
- Terapia cognitiva: Conceptos básicos y profundizaciónDe EverandTerapia cognitiva: Conceptos básicos y profundizaciónCalificación: 5 de 5 estrellas5/5 (1)
- Póngase En Forma Sin Salir De Su CasaDe EverandPóngase En Forma Sin Salir De Su CasaCalificación: 4.5 de 5 estrellas4.5/5 (4)
- Hipnoparto Y Parto Creativo: Cómo dar a luz con confianza y sin miedo Altravez del Método LeclaireDe EverandHipnoparto Y Parto Creativo: Cómo dar a luz con confianza y sin miedo Altravez del Método LeclaireCalificación: 4 de 5 estrellas4/5 (7)
- Sana tu Cuerpo, Calma tu Mente: Desintoxicar Hígado, Intestino Permeable, Salud Hormonal, Curación Emocional, Relajación, Ansiedad y Sanidad mental, Atención Plena, Psicoterapia y NutriciónDe EverandSana tu Cuerpo, Calma tu Mente: Desintoxicar Hígado, Intestino Permeable, Salud Hormonal, Curación Emocional, Relajación, Ansiedad y Sanidad mental, Atención Plena, Psicoterapia y NutriciónCalificación: 5 de 5 estrellas5/5 (4)
- Psiconeuroinmunología para la práctica clínicaDe EverandPsiconeuroinmunología para la práctica clínicaCalificación: 5 de 5 estrellas5/5 (4)
- Prescripción de ejercico físico para la saludDe EverandPrescripción de ejercico físico para la saludCalificación: 5 de 5 estrellas5/5 (1)
- Puntos gatillo y cadenas musculares funcionales en osteopatía y terapia manual (Bicolor)De EverandPuntos gatillo y cadenas musculares funcionales en osteopatía y terapia manual (Bicolor)Calificación: 4.5 de 5 estrellas4.5/5 (23)
- TDAH en Adultos. Cómo Reconocer y Tratar a un Adulto con TDAH en 30 Fáciles PasosDe EverandTDAH en Adultos. Cómo Reconocer y Tratar a un Adulto con TDAH en 30 Fáciles PasosCalificación: 4 de 5 estrellas4/5 (8)
- Programa de la psicoeducación de la ansiedad y entrenamiento en técnicas de relajación: Manual del TerapeutaDe EverandPrograma de la psicoeducación de la ansiedad y entrenamiento en técnicas de relajación: Manual del TerapeutaCalificación: 4 de 5 estrellas4/5 (8)
- Trauma, miedo y amor: Hacia una autonomía interior con la ayuda de las constelacionesDe EverandTrauma, miedo y amor: Hacia una autonomía interior con la ayuda de las constelacionesCalificación: 1 de 5 estrellas1/5 (1)
- La metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceDe EverandLa metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceCalificación: 5 de 5 estrellas5/5 (8)
- Manual de urología esencialDe EverandManual de urología esencialCalificación: 5 de 5 estrellas5/5 (1)
- El autismo: Reflexiones y pautas para comprenderlo y abordarloDe EverandEl autismo: Reflexiones y pautas para comprenderlo y abordarloCalificación: 4 de 5 estrellas4/5 (7)
- Aunque me cueste la vida: Las constelaciones familiares en casos de enfermedades crónicas y síntomas persistentesDe EverandAunque me cueste la vida: Las constelaciones familiares en casos de enfermedades crónicas y síntomas persistentesCalificación: 4.5 de 5 estrellas4.5/5 (9)
- Aprendizaje del examen clínico de los equinos, bovinos y caninosDe EverandAprendizaje del examen clínico de los equinos, bovinos y caninosCalificación: 4.5 de 5 estrellas4.5/5 (3)
- Psicoterapia breve con niños y adolescentesDe EverandPsicoterapia breve con niños y adolescentesCalificación: 4.5 de 5 estrellas4.5/5 (15)
- Alimentación y nutrición en dislipidemias, síndrome metabólico y enfermedad cardiovascularDe EverandAlimentación y nutrición en dislipidemias, síndrome metabólico y enfermedad cardiovascularCalificación: 5 de 5 estrellas5/5 (2)