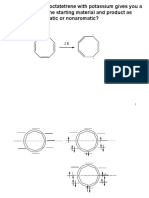
También podría gustarte
- Org Chap4 BirheDocumento42 páginasOrg Chap4 BirhebedadadenbalprosperityAún no hay calificaciones
- L2 Alkynes and AromaticsDocumento17 páginasL2 Alkynes and AromaticsCheng FuAún no hay calificaciones
- Chapter 13Documento24 páginasChapter 13abubakarabubakarbah563Aún no hay calificaciones
- Aromaticity & Aromatic Compounds IiDocumento63 páginasAromaticity & Aromatic Compounds Iidicoz diphaAún no hay calificaciones
- ORM-II TH EDocumento12 páginasORM-II TH EelonmuskonmoonAún no hay calificaciones
- Electrophilic Aromatic Substitution Reaction - by WWW - Learnengineering.inDocumento16 páginasElectrophilic Aromatic Substitution Reaction - by WWW - Learnengineering.inKripanshu KaushikAún no hay calificaciones
- Chapter 22-Benzos PDFDocumento29 páginasChapter 22-Benzos PDFSaskhia GomezAún no hay calificaciones
- 5.4.1 Arenes635464Documento6 páginas5.4.1 Arenes635464ArchitAún no hay calificaciones
- Aromatic HydrocarbonsDocumento18 páginasAromatic Hydrocarbonsapi-3734333Aún no hay calificaciones
- Alk EnesDocumento19 páginasAlk EnesFury GeorgeAún no hay calificaciones
- Intro To Organic Reactions CHM457Documento73 páginasIntro To Organic Reactions CHM457Zafrel ZaffAún no hay calificaciones
- UNG Aromatic 2Documento36 páginasUNG Aromatic 2Chha-fiidhAún no hay calificaciones
- Reactions of Benzene and Its DerivativeDocumento46 páginasReactions of Benzene and Its Derivativequyenda08hhaAún no hay calificaciones
- ReactionsDocumento114 páginasReactionshamdy solimanAún no hay calificaciones
- Goc 2 PDFDocumento16 páginasGoc 2 PDFAditya KadamAún no hay calificaciones
- 3 3 Revision Guide HalogenoalkanesDocumento5 páginas3 3 Revision Guide HalogenoalkanesmarieAún no hay calificaciones
- Aromatic Electrophilic Substitution ReactionsDocumento17 páginasAromatic Electrophilic Substitution ReactionsSai krishnaAún no hay calificaciones
- Naming HalogenoalkanesDocumento13 páginasNaming HalogenoalkanesAsma AkterAún no hay calificaciones
- Chapter 4Documento28 páginasChapter 4c4.arsyadAún no hay calificaciones
- "Recipe" For Consciousness: 1. Parietal Cortex 2. Frontal Cortex 3. Claustrum 4. Thalamus 5. Reticular Activating SystemDocumento62 páginas"Recipe" For Consciousness: 1. Parietal Cortex 2. Frontal Cortex 3. Claustrum 4. Thalamus 5. Reticular Activating SystemYupAún no hay calificaciones
- Electrophilic Aromatic Substitution + The Chemistry of BenzeneDocumento28 páginasElectrophilic Aromatic Substitution + The Chemistry of BenzeneTrescia Mae EstilloreAún no hay calificaciones
- 3.3 Revision Guide Halogenoalkanes AqaDocumento5 páginas3.3 Revision Guide Halogenoalkanes Aqashafiqur rahmanAún no hay calificaciones
- 16H Carbonyl PDFDocumento60 páginas16H Carbonyl PDFJose Erick Ortega ValenciaAún no hay calificaciones
- Lecture 2 - Reactions of Aromatic CompoundsDocumento164 páginasLecture 2 - Reactions of Aromatic CompoundsQutaiba IbrahimAún no hay calificaciones
- Polunuclear Hydrocarbon: Napthalene: As Per PCI Curriculum Pharmaceutical Chemistry-II Second Year B. Pharmacy (Sem-III)Documento21 páginasPolunuclear Hydrocarbon: Napthalene: As Per PCI Curriculum Pharmaceutical Chemistry-II Second Year B. Pharmacy (Sem-III)Ronak Modi100% (1)
- Chemistry of Benzene: Electrophilic Aromatic SubstitutionDocumento80 páginasChemistry of Benzene: Electrophilic Aromatic Substitution張湧浩100% (1)
- Polunuclear Hydrocarbon: Napthalene: As Per PCI Curriculum Pharmaceutical Chemistry-II Second Year B. Pharmacy (Sem-III)Documento22 páginasPolunuclear Hydrocarbon: Napthalene: As Per PCI Curriculum Pharmaceutical Chemistry-II Second Year B. Pharmacy (Sem-III)Ronak ModiAún no hay calificaciones
- Sebatian AromatikDocumento100 páginasSebatian AromatikGanthimathi SugumaranAún no hay calificaciones
- ORM-II Theory+exercise+ Answer PDFDocumento58 páginasORM-II Theory+exercise+ Answer PDFGOURISH AGRAWALAún no hay calificaciones
- JB Chemical Ideas 12point3arenesDocumento9 páginasJB Chemical Ideas 12point3arenesOCRChemistrySaltersAún no hay calificaciones
- Addition Reactions of AlkynesDocumento26 páginasAddition Reactions of Alkynesaggelisgeorge8546Aún no hay calificaciones
- Aromatic Notes 2 PDFDocumento6 páginasAromatic Notes 2 PDFChris_Barber09100% (1)
- Aromatic Electrophilic SubstitutionDocumento71 páginasAromatic Electrophilic SubstitutionsridharancAún no hay calificaciones
- Benceno SEA PDFDocumento6 páginasBenceno SEA PDFcarolinehwangAún no hay calificaciones
- Application of Resonance EffectDocumento6 páginasApplication of Resonance EffectShanky HaryanviAún no hay calificaciones
- Physical Organic Chemistry Chapter ThreeDocumento40 páginasPhysical Organic Chemistry Chapter ThreeMULUKEN TILAHUNAún no hay calificaciones
- Chemistry 202: Reactions of Alkenes, Part 1Documento28 páginasChemistry 202: Reactions of Alkenes, Part 1CindyAún no hay calificaciones
- Chapter 5 Elimination Reaction - 2016Documento19 páginasChapter 5 Elimination Reaction - 2016Syuhadah NoordinAún no hay calificaciones
- Alkene: The "Double Bonds" in A Benzene Ring Do Not React Like OthersDocumento18 páginasAlkene: The "Double Bonds" in A Benzene Ring Do Not React Like OthersPenny LowAún no hay calificaciones
- Two Types of Hydrocarbons: Open in Desktop App. Aromatic Compounds (Arene)Documento11 páginasTwo Types of Hydrocarbons: Open in Desktop App. Aromatic Compounds (Arene)Freya SawAún no hay calificaciones
- Lesson 8 Reactions of HalogenoalkanesDocumento15 páginasLesson 8 Reactions of Halogenoalkanesdela2Aún no hay calificaciones
- Chapter 3Documento28 páginasChapter 3c4.arsyadAún no hay calificaciones
- Two Types of Hydrocarbons: Aromatic Compounds (Arene)Documento11 páginasTwo Types of Hydrocarbons: Aromatic Compounds (Arene)Freya SawAún no hay calificaciones
- Bab Benzene and Aromaticity+ (II)Documento48 páginasBab Benzene and Aromaticity+ (II)ghaida farmasiAún no hay calificaciones
- Lec07 Notes Jan23Documento7 páginasLec07 Notes Jan23KataAún no hay calificaciones
- Hydrocarbon Compounds: AromaticDocumento49 páginasHydrocarbon Compounds: AromaticUMMU MARDHIAH ABDUL HALIMAún no hay calificaciones
- Reactivity and Mechanism: Presented By: Dr. Faisal Almalki (Male)Documento30 páginasReactivity and Mechanism: Presented By: Dr. Faisal Almalki (Male)battal eduAún no hay calificaciones
- 22 Eas Revision Notes QuizrrDocumento52 páginas22 Eas Revision Notes QuizrrMONEY ALLAún no hay calificaciones
- Indoles and IsoindolesDocumento16 páginasIndoles and IsoindolesSylvester AsareAún no hay calificaciones
- Aromatic Compound Theory - EDocumento23 páginasAromatic Compound Theory - Ethinkiit100% (1)
- Chapter 12Documento23 páginasChapter 12Hải NguyễnAún no hay calificaciones
- AS Level - AlkenesDocumento17 páginasAS Level - AlkenesRegine BalagtasAún no hay calificaciones
- Things To Remember Only Alc Phe 2022-23Documento17 páginasThings To Remember Only Alc Phe 2022-23poornaAún no hay calificaciones
- Summary Sheet: Two Key Concepts For Nucleophilic Substitution On CarbonylsDocumento1 páginaSummary Sheet: Two Key Concepts For Nucleophilic Substitution On CarbonylsChitKoAún no hay calificaciones
- Aromatic ChemistryDocumento16 páginasAromatic ChemistrysimbaleoAún no hay calificaciones
- Hyperconjugation - Dr. Akshay ShuklaDocumento26 páginasHyperconjugation - Dr. Akshay ShuklawaqasAún no hay calificaciones
- Che232 L10Documento3 páginasChe232 L10rutasojaneAún no hay calificaciones
- Chemsheets A2 1025 Reactions of AromaticsDocumento5 páginasChemsheets A2 1025 Reactions of AromaticsEbtihal AlharthiAún no hay calificaciones
- Organometallic Chemistry: Plenary Lectures Presented at the Fourth International Conference on Organometallic ChemistryDe EverandOrganometallic Chemistry: Plenary Lectures Presented at the Fourth International Conference on Organometallic ChemistryF. G. A. StoneAún no hay calificaciones
- XXIVth International Congress of Pure and Applied Chemistry: Plenary and Main Section Lectures Presented at Hamburg, Federal Republic of Germany, 2–8 September 1973De EverandXXIVth International Congress of Pure and Applied Chemistry: Plenary and Main Section Lectures Presented at Hamburg, Federal Republic of Germany, 2–8 September 1973Aún no hay calificaciones
- The Genomics of Speciation With Gene-FlowDocumento10 páginasThe Genomics of Speciation With Gene-FlowJSGIAún no hay calificaciones
- Pinker - The Past-Tense DebateDocumento8 páginasPinker - The Past-Tense DebateJSGIAún no hay calificaciones
- A New Technique For Identifying Sequence HeterochronyDocumento11 páginasA New Technique For Identifying Sequence HeterochronyJSGIAún no hay calificaciones
- Heterochrony Revisited - The Evolution of Developmental SequencesDocumento18 páginasHeterochrony Revisited - The Evolution of Developmental SequencesJSGIAún no hay calificaciones
- Pinker - Toward A Consilient Study of LiteratureDocumento17 páginasPinker - Toward A Consilient Study of LiteratureJSGIAún no hay calificaciones
- Pinker - What It Means To Be HumanDocumento1 páginaPinker - What It Means To Be HumanJSGIAún no hay calificaciones
- Transposable Elements DynamicsDocumento14 páginasTransposable Elements DynamicsJSGIAún no hay calificaciones
- Pinker Etal 2007Documento6 páginasPinker Etal 2007ecoAún no hay calificaciones
- Neuronal Cell Death in Huntington's Disease - A Potential Role For Dopamine (2000)Documento7 páginasNeuronal Cell Death in Huntington's Disease - A Potential Role For Dopamine (2000)JSGIAún no hay calificaciones
- Adult Neurogenesis - From Precursors To Network and Physiology (2005)Documento49 páginasAdult Neurogenesis - From Precursors To Network and Physiology (2005)JSGIAún no hay calificaciones
- The Nearly Neutral Theory of Molecular Evolution (1992)Documento26 páginasThe Nearly Neutral Theory of Molecular Evolution (1992)JSGIAún no hay calificaciones
- DcdennettDocumento4 páginasDcdennettJSGIAún no hay calificaciones
- Long-Term Potentiation & Memory HealthDocumento52 páginasLong-Term Potentiation & Memory HealthJSGIAún no hay calificaciones
- Physiology and Neurobiology of Stress and Adaptation - Central Role of The BrainDocumento33 páginasPhysiology and Neurobiology of Stress and Adaptation - Central Role of The BrainJSGIAún no hay calificaciones
- The Historical Roots of Our Ecological Crisis, Lynn WhiteDocumento7 páginasThe Historical Roots of Our Ecological Crisis, Lynn WhiteJSGI100% (2)
- Unit 4 StudyDocumento1 páginaUnit 4 StudyJSGIAún no hay calificaciones
- Ch20 Hartwell4eDocumento42 páginasCh20 Hartwell4eJSGIAún no hay calificaciones
- Chapter 5Documento19 páginasChapter 5JSGIAún no hay calificaciones
- Parfit - Why Does The Universe ExistDocumento2 páginasParfit - Why Does The Universe ExistRickard Vester SmetanaAún no hay calificaciones
- Answers To End-Of-Chapter QuestionsDocumento8 páginasAnswers To End-Of-Chapter Questionsluizdr100% (1)
- Atm Benzene TopicDocumento31 páginasAtm Benzene Topicpratik kumarAún no hay calificaciones
- Topic Exploration Pack Reactions of Phenols: Instructions and Answers For TeachersDocumento31 páginasTopic Exploration Pack Reactions of Phenols: Instructions and Answers For TeachersCamille deanAún no hay calificaciones
- BenzeneDocumento54 páginasBenzeneMike EzioAún no hay calificaciones
- Aromatic Compounds and Uses of BenzeneDocumento44 páginasAromatic Compounds and Uses of BenzenePaarijat DubeyAún no hay calificaciones
- Expt 7 Classification Tests For HydrocarbonsDocumento10 páginasExpt 7 Classification Tests For Hydrocarbonssean goAún no hay calificaciones
- Peter Atkins Julio de Paula Ron Friedman Physical Chemistry Quanta (0256-0306)Documento51 páginasPeter Atkins Julio de Paula Ron Friedman Physical Chemistry Quanta (0256-0306)Administracion OTIC IVICAún no hay calificaciones
- Faculty of Chemical Engineering: ObjectiveDocumento5 páginasFaculty of Chemical Engineering: ObjectiveMohd RusyaidiAún no hay calificaciones
- ICT Mumbai B.Chem - Engg SyllabusDocumento41 páginasICT Mumbai B.Chem - Engg SyllabusAniruddha ShidhayeAún no hay calificaciones
- Resonance StructuresDocumento4 páginasResonance StructuresSmyra100% (1)
- AromaticityDocumento24 páginasAromaticityCSH Study WebAún no hay calificaciones
- CAPE Chemistry Mock Exams U2 2015Documento19 páginasCAPE Chemistry Mock Exams U2 2015Nicholas CharlesAún no hay calificaciones
- (Advances in Agronomy Volume 141) Donald L. Sparks (Eds.) - Academic Press (2017)Documento279 páginas(Advances in Agronomy Volume 141) Donald L. Sparks (Eds.) - Academic Press (2017)Luis Fernando Guayllas PomaAún no hay calificaciones
- Heterocyclic Compounds 1 فصل ثاني مرحلة ثانية مادة العضويةDocumento32 páginasHeterocyclic Compounds 1 فصل ثاني مرحلة ثانية مادة العضويةLootus FlowerAún no hay calificaciones
- CH13 Hydrocarbons Shobhit NirwanDocumento58 páginasCH13 Hydrocarbons Shobhit NirwanpujaAún no hay calificaciones
- Aroma TikDocumento17 páginasAroma TikStarvilla HAún no hay calificaciones
- Aromatic Chemistry4.6 NotesDocumento674 páginasAromatic Chemistry4.6 Noteskudec2008Aún no hay calificaciones
- ChemsketchDocumento9 páginasChemsketchTubagus SinggihAún no hay calificaciones
- CHM457 Lab 5 PDFDocumento6 páginasCHM457 Lab 5 PDFsuraini100% (1)
- The Journal of Chemical Physics Volume 20 Issue 4 1952 (Doi 10.1063 - 1.1700516) McClure, Donald S. - Spin-Orbit Interaction in Aromatic MoleculesDocumento6 páginasThe Journal of Chemical Physics Volume 20 Issue 4 1952 (Doi 10.1063 - 1.1700516) McClure, Donald S. - Spin-Orbit Interaction in Aromatic MoleculesJustin MdAún no hay calificaciones
- CR 000442 TDocumento34 páginasCR 000442 TMys GenieAún no hay calificaciones
- Organic ChemistryDocumento864 páginasOrganic Chemistryforumchemitry100% (1)
- Aromaticity: Reactions of Benzene and Substituted Benzenes: Essential Organic Chemistry (Bruice)Documento41 páginasAromaticity: Reactions of Benzene and Substituted Benzenes: Essential Organic Chemistry (Bruice)tyron9520Aún no hay calificaciones
- Heterocyclic Compounds MergedDocumento158 páginasHeterocyclic Compounds MergedBisma ShafiqAún no hay calificaciones
- Science 9: The Chemistry of CarbonDocumento22 páginasScience 9: The Chemistry of CarbonAnastacia Anne Eva CambaAún no hay calificaciones
- Chapter3 HydrocarbonsDocumento19 páginasChapter3 Hydrocarbonssyaz lianaAún no hay calificaciones
- Petrochem 10 - SEM 1 12-13Documento40 páginasPetrochem 10 - SEM 1 12-13Saifuddin AzizAún no hay calificaciones
- 1976 FixationDocumento9 páginas1976 FixationMark EvansAún no hay calificaciones
- Bicycloalkane NomenclatureDocumento41 páginasBicycloalkane NomenclatureVăn Nam LêAún no hay calificaciones
- Process Engineering and Design: A Solvent-Resid Phase Diagram For Tracking Resid ConversionDocumento7 páginasProcess Engineering and Design: A Solvent-Resid Phase Diagram For Tracking Resid ConversionVinodh KumarAún no hay calificaciones