También podría gustarte
- Casos Practicos ResueltosDocumento18 páginasCasos Practicos ResueltosJose Manayay Sanchez70% (10)
- Testículos ausentes o malposicionados (criptorquidia, ectopia, agenesiaDocumento35 páginasTestículos ausentes o malposicionados (criptorquidia, ectopia, agenesiaJosè Jesùs Rincòn Sànchez100% (1)
- Curso Taller Upsjb 2021 LimaDocumento22 páginasCurso Taller Upsjb 2021 LimaDerivan José Peña zarateAún no hay calificaciones
- SolasDocumento70 páginasSolasCrystal RiveraAún no hay calificaciones
- receipt-01-20212-0-1-1-1-ICA-AN-10-20202104606-NU-Publicidad AnimadaDocumento1 páginareceipt-01-20212-0-1-1-1-ICA-AN-10-20202104606-NU-Publicidad AnimadaDerivan José Peña zarateAún no hay calificaciones
- Xiaomi PDocumento8 páginasXiaomi PDerivan José Peña zarateAún no hay calificaciones
- 3ra. Sesion-2019 - 20190906101427Documento54 páginas3ra. Sesion-2019 - 20190906101427Derivan José Peña zarateAún no hay calificaciones
- Empleo y La InflaciónDocumento16 páginasEmpleo y La InflaciónDerivan José Peña zarateAún no hay calificaciones
- Fichas Descriptivas de 52 PDFDocumento82 páginasFichas Descriptivas de 52 PDFargos1301Aún no hay calificaciones
- Mapa - Peña ZarateDocumento1 páginaMapa - Peña ZarateDerivan José Peña zarateAún no hay calificaciones
- Teoria General de SistemasDocumento7 páginasTeoria General de SistemasDerivan José Peña zarateAún no hay calificaciones
- Flo RRRRRDocumento8 páginasFlo RRRRRDerivan José Peña zarateAún no hay calificaciones
- Técnicas y Métodos Creativos Aplicados A La Conceptualización Del DiseñoDocumento4 páginasTécnicas y Métodos Creativos Aplicados A La Conceptualización Del DiseñoJohn PretelAún no hay calificaciones
- Ley30230 Defensa Posesoria Extrajudicial Articulo 920 Codigo Civil PDFDocumento18 páginasLey30230 Defensa Posesoria Extrajudicial Articulo 920 Codigo Civil PDFMariana MuñozAún no hay calificaciones
- Arte Investigacion FormativaDocumento44 páginasArte Investigacion FormativaDerivan José Peña zarateAún no hay calificaciones
- Universidad Nacional Del Centro Del PerúDocumento154 páginasUniversidad Nacional Del Centro Del PerúDerivan José Peña zarateAún no hay calificaciones
- Ejercicio FONOTELDocumento2 páginasEjercicio FONOTELFrank Piñin AtoAún no hay calificaciones
- Derian Peña Zarate - Cadena de Valor de Las Marcas D EmodaDocumento1 páginaDerian Peña Zarate - Cadena de Valor de Las Marcas D EmodaDerivan José Peña zarateAún no hay calificaciones
- Entrevista Aun DocenteDocumento1 páginaEntrevista Aun DocenteDerivan José Peña zarateAún no hay calificaciones
- Costos producción tablaDocumento2 páginasCostos producción tablaDerivan José Peña zarate100% (1)
- 1Documento1 página1Derivan José Peña zarateAún no hay calificaciones
- Entrevista Aun DocenteDocumento1 páginaEntrevista Aun DocenteDerivan José Peña zarateAún no hay calificaciones
- Grafico EconomiaDocumento2 páginasGrafico EconomiaDerivan José Peña zarateAún no hay calificaciones
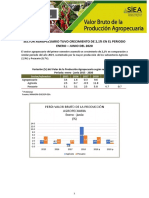
- VBP Junio 2020Documento5 páginasVBP Junio 2020Derivan José Peña zarateAún no hay calificaciones
- CapDocumento3 páginasCapDerivan José Peña zarateAún no hay calificaciones
- Ley N 0001Documento2 páginasLey N 0001Derivan José Peña zarateAún no hay calificaciones
- Regímenes Tributarios 2020 - SunatDocumento3 páginasRegímenes Tributarios 2020 - SunatGiancarlo CordovaAún no hay calificaciones
- FONDEMDocumento2 páginasFONDEMDerivan José Peña zarateAún no hay calificaciones
- 3.2 Lectura El Libro MayorDocumento2 páginas3.2 Lectura El Libro MayorHumberto FloresAún no hay calificaciones
- Código de BarrasDocumento1 páginaCódigo de BarrasDerivan José Peña zarateAún no hay calificaciones
- Cronograma de Actividades Académicas 2020 - 20 de Abril - VRA - ICADocumento2 páginasCronograma de Actividades Académicas 2020 - 20 de Abril - VRA - ICADerivan José Peña zarateAún no hay calificaciones
- Simulidos en HuanucoDocumento5 páginasSimulidos en HuanucoAlan EspinozaAún no hay calificaciones
- Proyecto Control Prevencion PediculosisDocumento98 páginasProyecto Control Prevencion PediculosisElis RamirezAún no hay calificaciones
- Matriz de IPEVR Empresa Arepas El Buen Gusto S.ADocumento89 páginasMatriz de IPEVR Empresa Arepas El Buen Gusto S.AJuan Carlos DominguezAún no hay calificaciones
- Clasificación Desechos Acuerdo Ministerial 323Documento4 páginasClasificación Desechos Acuerdo Ministerial 323karla velarde80% (5)
- Definicion Del Paciente Con Necesidades PaliativasDocumento24 páginasDefinicion Del Paciente Con Necesidades PaliativasCristopher PonceAún no hay calificaciones
- Exploración física general de cabeza a piesDocumento15 páginasExploración física general de cabeza a piesJohnnyCarbajalGuzmanAún no hay calificaciones
- 6º Octubre EjerciciosDocumento43 páginas6º Octubre EjerciciosLETICIA RENDON50% (2)
- 1 Primeros Auxilios Evaluación PrimariaDocumento50 páginas1 Primeros Auxilios Evaluación PrimariaAlexAún no hay calificaciones
- AZAFRÁNnnDocumento21 páginasAZAFRÁNnnJulian RamirezAún no hay calificaciones
- Ensayo Covid19-Vasquez WendyDocumento5 páginasEnsayo Covid19-Vasquez WendyWendy Viviana Vasquez DelgadoAún no hay calificaciones
- Graficos de Silvia TesisDocumento16 páginasGraficos de Silvia TesisevelynAún no hay calificaciones
- Reporte Rayos X TyrionDocumento4 páginasReporte Rayos X TyrionAlex EstradaAún no hay calificaciones
- Ansiedad y Convulsiones Histéricas FinalDocumento16 páginasAnsiedad y Convulsiones Histéricas FinalBeatriz RodriguezAún no hay calificaciones
- Calendarización Del Año Escolar 2021Documento3 páginasCalendarización Del Año Escolar 2021Luz VargasAún no hay calificaciones
- Salud BucalDocumento7 páginasSalud BucalQUISPE GOMEZ LOURDES ISAYDAAún no hay calificaciones
- Simulacro Examen Oposiciones Técnico Analisis ClinicosDocumento38 páginasSimulacro Examen Oposiciones Técnico Analisis ClinicosJesica PerezAún no hay calificaciones
- Crianza ExtensivaDocumento62 páginasCrianza ExtensivaPlinio SeRnaAún no hay calificaciones
- Tuberculosis infantil: diagnóstico y tratamientoDocumento17 páginasTuberculosis infantil: diagnóstico y tratamientoMilagros Vela SalazarAún no hay calificaciones
- Proteus, Shigella, E ColiiDocumento43 páginasProteus, Shigella, E ColiiAlexis Zárate de HernándezAún no hay calificaciones
- Conocimiento y uso de plantas medicinales por estudiantes universitariosDocumento15 páginasConocimiento y uso de plantas medicinales por estudiantes universitariosSarita QuinteroAún no hay calificaciones
- Covid-19 en Honduras: análisis comparativo de impacto socioeconómicoDocumento28 páginasCovid-19 en Honduras: análisis comparativo de impacto socioeconómicosolo_alejo93Aún no hay calificaciones
- Pancreatitis AgudaDocumento11 páginasPancreatitis AgudaStefany Casas FloresAún no hay calificaciones
- Determinación Del Grupo Sanguíneo y Factor RHDocumento2 páginasDeterminación Del Grupo Sanguíneo y Factor RHSoy AraAún no hay calificaciones
- Cuadernillo de IdentificaciónDocumento17 páginasCuadernillo de Identificaciónluis vicunaAún no hay calificaciones
- Taller de Ausentismo LaboralDocumento6 páginasTaller de Ausentismo LaboralDANIEL ANDREWSAún no hay calificaciones
- Factores SOAT procedimientos oftalmológicosDocumento148 páginasFactores SOAT procedimientos oftalmológicosMONIKPATRICIA RUBIANO ORDOÑEZAún no hay calificaciones
- Sistema óseo, muscular y nervioso: prueba ciencias naturalesDocumento5 páginasSistema óseo, muscular y nervioso: prueba ciencias naturalesGabriel Jara MuñozAún no hay calificaciones
- Trabajo MecanografíaDocumento12 páginasTrabajo Mecanografíalaies561Aún no hay calificaciones
- Ictericia NeonatalDocumento30 páginasIctericia NeonatalRaquel GarcíaAún no hay calificaciones