También podría gustarte
- Ebselen, A Seleno-Organic Antioxidant, Is Neuroprotective After Embolic Strokes in RabbitsDocumento7 páginasEbselen, A Seleno-Organic Antioxidant, Is Neuroprotective After Embolic Strokes in RabbitsZahra NisaAún no hay calificaciones
- Ebselen, A Seleno-Organic Antioxidant, Is Neuroprotective After Embolic Strokes in RabbitsDocumento7 páginasEbselen, A Seleno-Organic Antioxidant, Is Neuroprotective After Embolic Strokes in RabbitsZahra NisaAún no hay calificaciones
- JurnalDocumento8 páginasJurnalSyifa MawaddahAún no hay calificaciones
- Neuroprotective Effect of Curcumin in An Experimental Rat Model of Subarachnoid HemorrhageDocumento20 páginasNeuroprotective Effect of Curcumin in An Experimental Rat Model of Subarachnoid HemorrhageShalim BagusAún no hay calificaciones
- 16 Jagathidevi EtalDocumento5 páginas16 Jagathidevi EtaleditorijmrhsAún no hay calificaciones
- Am J Physiol Regul Integr Comp Physiol 2001 - BadrDocumento6 páginasAm J Physiol Regul Integr Comp Physiol 2001 - BadrTony LeeAún no hay calificaciones
- Nai Et Al 2017 Protective Effect of Astaxanthin On Acute Cerebral Infarction in RatsDocumento8 páginasNai Et Al 2017 Protective Effect of Astaxanthin On Acute Cerebral Infarction in RatsMennatallah AliAún no hay calificaciones
- Phytotherapy Research Volume 24 Issue 3 2010 (Doi 10.1002/ptr.2982) Hyung-In Moon Ill-Min Chung Su-Hyun SEO Eun-Young Kang - Protective Effects of 3 - Deoxy-4-O-methylepiDocumento3 páginasPhytotherapy Research Volume 24 Issue 3 2010 (Doi 10.1002/ptr.2982) Hyung-In Moon Ill-Min Chung Su-Hyun SEO Eun-Young Kang - Protective Effects of 3 - Deoxy-4-O-methylepiFaradita NindyasariAún no hay calificaciones
- BMC Clinical Pathology: Protection of Early Phase Hepatic Ischemia-Reperfusion Injury by Cholinergic AgonistsDocumento13 páginasBMC Clinical Pathology: Protection of Early Phase Hepatic Ischemia-Reperfusion Injury by Cholinergic AgonistsPrachi KariaAún no hay calificaciones
- Ni Hms 406767Documento19 páginasNi Hms 406767Alex BuruianaAún no hay calificaciones
- Bioorganic & Medicinal ChemistryDocumento9 páginasBioorganic & Medicinal ChemistryVinayak KhairnarAún no hay calificaciones
- Cerebrolysin Increases Recovery and Decreases Inflammation in An Autoimmune Model of Autoimmune EncephalitisDocumento15 páginasCerebrolysin Increases Recovery and Decreases Inflammation in An Autoimmune Model of Autoimmune EncephalitisAlin MihaiAún no hay calificaciones
- ArtículoDocumento10 páginasArtículoKaty RamírezAún no hay calificaciones
- Dhami Et Al-2019-Journal of Neurochemistry PDFDocumento18 páginasDhami Et Al-2019-Journal of Neurochemistry PDFJawad A. KhanAún no hay calificaciones
- Epigallocatechin-3-Gallate Protects Against Neuronal Cell Death and Improves Cerebral Function After Traumatic Brain Injury in RatsDocumento10 páginasEpigallocatechin-3-Gallate Protects Against Neuronal Cell Death and Improves Cerebral Function After Traumatic Brain Injury in RatsManu PatilAún no hay calificaciones
- α‐lipoic acid protects against cerebral ischemia - reperfusion‐induced injury in ratsDocumento7 páginasα‐lipoic acid protects against cerebral ischemia - reperfusion‐induced injury in ratsranu anggaraAún no hay calificaciones
- Jia Yin, Haiying Li, Chengjie Meng, Dongdong Chen, Zhouqing Chen, Yibin Wang, Zhong Wang, Gang ChenDocumento12 páginasJia Yin, Haiying Li, Chengjie Meng, Dongdong Chen, Zhouqing Chen, Yibin Wang, Zhong Wang, Gang ChenkasabeAún no hay calificaciones
- Journal of Ethnopharmacology: Anti-Amnesic Effect of Chong-Myung-Tang On Scopolamine-Induced Memory Impairments in MiceDocumento5 páginasJournal of Ethnopharmacology: Anti-Amnesic Effect of Chong-Myung-Tang On Scopolamine-Induced Memory Impairments in MiceSujith KuttanAún no hay calificaciones
- 08 Chapter 4Documento19 páginas08 Chapter 4annisapritasAún no hay calificaciones
- NeuroDocumento9 páginasNeuromamun31Aún no hay calificaciones
- Proceedings of The Anaesthetic Research Society Meeting: AbstractsDocumento13 páginasProceedings of The Anaesthetic Research Society Meeting: AbstractsrYanDYAún no hay calificaciones
- Beneficial Effect of Cyclosporine A On Traumatic Hemorrhagic ShockDocumento12 páginasBeneficial Effect of Cyclosporine A On Traumatic Hemorrhagic ShockJoecoAún no hay calificaciones
- Curcumin Provides Neuroprotection After Spinal Cord Injury: Ó 2009 Elsevier Inc. All Rights ReservedDocumento10 páginasCurcumin Provides Neuroprotection After Spinal Cord Injury: Ó 2009 Elsevier Inc. All Rights ReservedanthonyjoelleeAún no hay calificaciones
- Biocompatibility Evaluation of Tramadol Loaded Nanoparticulate SystemsDocumento2 páginasBiocompatibility Evaluation of Tramadol Loaded Nanoparticulate SystemsVeronica NedelcuAún no hay calificaciones
- Neurology & NeurophysiologyDocumento6 páginasNeurology & NeurophysiologyRakesh KumarAún no hay calificaciones
- BMC Complementary and Alternative MedicineDocumento12 páginasBMC Complementary and Alternative MedicineRudra Abhishek KumarAún no hay calificaciones
- Protective Eff Ects of Ginkgo Biloba Extract (EGB 761) On Astrocytes of Rat Hippocampus After Exposure With ScopolamineDocumento5 páginasProtective Eff Ects of Ginkgo Biloba Extract (EGB 761) On Astrocytes of Rat Hippocampus After Exposure With Scopolamineaurelio.arae713Aún no hay calificaciones
- Bioorganic & Medicinal ChemistryDocumento14 páginasBioorganic & Medicinal ChemistryAulia Fatmiyatun El-FalesyAún no hay calificaciones
- Articulo para Expo TraducirDocumento6 páginasArticulo para Expo TraducirViki G PAún no hay calificaciones
- UntitledDocumento7 páginasUntitledTuấn Nguyen AnhAún no hay calificaciones
- Oxidative Stress Is The Primary Event: Effects of Ethanol Consumption in BrainDocumento6 páginasOxidative Stress Is The Primary Event: Effects of Ethanol Consumption in BrainMokhammad Faisol AbdullahAún no hay calificaciones
- Escitalopram Oxalate, A Selective Serotonin Reuptake Inhibitor, Exhibits Cytotoxic and Apoptotic Effects in Glioma C6 CellsDocumento6 páginasEscitalopram Oxalate, A Selective Serotonin Reuptake Inhibitor, Exhibits Cytotoxic and Apoptotic Effects in Glioma C6 CellsMary FallAún no hay calificaciones
- Zheng 2013Documento10 páginasZheng 2013Aldo ValenzuelaAún no hay calificaciones
- Effect Mangosteen ExtractDocumento19 páginasEffect Mangosteen ExtractAndi ZulkarnaeAún no hay calificaciones
- Pluripotent Anti-Inflammatory Immunomodulatory Effects of Papaverine Against Cerebral Ischemic-Reperfusion InjuryDocumento23 páginasPluripotent Anti-Inflammatory Immunomodulatory Effects of Papaverine Against Cerebral Ischemic-Reperfusion InjuryDragomir MirunaAún no hay calificaciones
- Ceftriaxone Therapy Attenuates Brain Trauma in Rats by Affecting Glutamate Transporters and Neuroinflammation and Not by Its Antibacterial EffectsDocumento14 páginasCeftriaxone Therapy Attenuates Brain Trauma in Rats by Affecting Glutamate Transporters and Neuroinflammation and Not by Its Antibacterial EffectsMuhammad Reza FirdausAún no hay calificaciones
- Pan 2015Documento13 páginasPan 2015Fran CiAún no hay calificaciones
- Effect of Melatonin On Blood Pressure and Nitric Oxide Generation in Rats With Metabolic SyndromeDocumento8 páginasEffect of Melatonin On Blood Pressure and Nitric Oxide Generation in Rats With Metabolic SyndromeExcelsis Deo SombolinggiAún no hay calificaciones
- Ngu Gia Bi Gai-Acanthopanax SenticosusDocumento6 páginasNgu Gia Bi Gai-Acanthopanax SenticosusMai Anh NguyễnAún no hay calificaciones
- Melatonina (2006) Cheung R, Safety IV RatasDocumento7 páginasMelatonina (2006) Cheung R, Safety IV RatasAlumno del Doctorado FarmacologiaAún no hay calificaciones
- Cardioproteccion Frente Isquemia 2008Documento8 páginasCardioproteccion Frente Isquemia 2008cumbredinAún no hay calificaciones
- Ajassp 2010 480 485Documento6 páginasAjassp 2010 480 485Mehmet BerközAún no hay calificaciones
- Astrocytic Expression of CTMP Following An Excitotoxic Lesion in The Mouse HippocampusDocumento8 páginasAstrocytic Expression of CTMP Following An Excitotoxic Lesion in The Mouse HippocampusSergeat18BAún no hay calificaciones
- 1percent EtOH Drinking-Water Lowers IGF-1 Insulin Glucose All Good But 2percent EtOH Not So Mice 2017Documento8 páginas1percent EtOH Drinking-Water Lowers IGF-1 Insulin Glucose All Good But 2percent EtOH Not So Mice 2017Gál Bence SzabóAún no hay calificaciones
- Ketogenic Diet Decreases Oxidative StressDocumento11 páginasKetogenic Diet Decreases Oxidative StressHocus PocusAún no hay calificaciones
- Comparizon of K-X and IsofluraneDocumento24 páginasComparizon of K-X and IsofluraneIgnacio BarbieriAún no hay calificaciones
- Comparison of The Spinal Neuropathic Pain Induced by Intraspinal Injection of N-Methyl-D-Aspartate and Quisquate in RatsDocumento6 páginasComparison of The Spinal Neuropathic Pain Induced by Intraspinal Injection of N-Methyl-D-Aspartate and Quisquate in Ratswicak_spAún no hay calificaciones
- The Effect of Dexpanthenol On Ototoxicity Induced by CisplatinDocumento7 páginasThe Effect of Dexpanthenol On Ototoxicity Induced by CisplatinNoval ArdianAún no hay calificaciones
- Jurding NeurologiDocumento11 páginasJurding NeurologiHasya KinasihAún no hay calificaciones
- AgNPs Paper 5Documento10 páginasAgNPs Paper 5VILEOLAGOLDAún no hay calificaciones
- New AED Lacosamide Shows Efficacy for Partial SeizuresDocumento7 páginasNew AED Lacosamide Shows Efficacy for Partial SeizuresRAHULAún no hay calificaciones
- JPharmacolPharmacother213-2966031 004926Documento4 páginasJPharmacolPharmacother213-2966031 004926David LittleAún no hay calificaciones
- 0708 0564 PDFDocumento18 páginas0708 0564 PDFRichard StephenAún no hay calificaciones
- Ijbms 19 800Documento4 páginasIjbms 19 800wineniAún no hay calificaciones
- The Effect of Confinement Stress On The Immune System An Endocrinology Laboratory Exercise DemonstratingDocumento5 páginasThe Effect of Confinement Stress On The Immune System An Endocrinology Laboratory Exercise DemonstratingArun Kumar N SAún no hay calificaciones
- The Effect of Repeated Exposure To Ethanol On Pre-Existing Fear Memories in RatsDocumento8 páginasThe Effect of Repeated Exposure To Ethanol On Pre-Existing Fear Memories in RatsAmanda MageskiAún no hay calificaciones
- Open Field - FarmaciaDocumento11 páginasOpen Field - Farmaciavlad_văluAún no hay calificaciones
- NIH Public AccessDocumento26 páginasNIH Public AccessEmy Noviana SandyAún no hay calificaciones
- Jurnal Untuk Soal Pak WYDocumento11 páginasJurnal Untuk Soal Pak WYPitra CharesnaAún no hay calificaciones
- 12.4.09 Laudate Spinal LesionsDocumento31 páginas12.4.09 Laudate Spinal LesionsDrGasnasAún no hay calificaciones
- ch15 APR LectureDocumento60 páginasch15 APR LectureDrGasnasAún no hay calificaciones
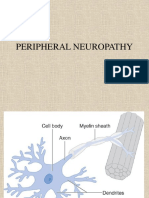
- Peripheral NeuropathyDocumento48 páginasPeripheral NeuropathyDrGasnasAún no hay calificaciones
- Chapter 16 LizDocumento58 páginasChapter 16 LizDrGasnasAún no hay calificaciones
- 9 AnsDocumento25 páginas9 AnsDrGasnasAún no hay calificaciones
- Lec. 2 - Autonomic Nervous SystemDocumento26 páginasLec. 2 - Autonomic Nervous SystemDrGasnasAún no hay calificaciones
- Autonomic Nervous SystemDocumento48 páginasAutonomic Nervous SystemDrGasnasAún no hay calificaciones
- Differentiating UMN and LMN LesionsDocumento4 páginasDifferentiating UMN and LMN LesionsDrGasnasAún no hay calificaciones
- NIH Public Access: Author ManuscriptDocumento18 páginasNIH Public Access: Author ManuscriptDrGasnasAún no hay calificaciones
- Perspective of Synaptic Protection After Post-Infarction Treatment With StatinsDocumento9 páginasPerspective of Synaptic Protection After Post-Infarction Treatment With StatinsDrGasnasAún no hay calificaciones
- Encephalitiscia3 151011085926 Lva1 App6892Documento43 páginasEncephalitiscia3 151011085926 Lva1 App6892DrGasnasAún no hay calificaciones
- Role of GABA Plasticity in StrokeDocumento3 páginasRole of GABA Plasticity in StrokeDrGasnasAún no hay calificaciones
- In Ammatory Mechanisms in Ischemic Stroke PDFDocumento11 páginasIn Ammatory Mechanisms in Ischemic Stroke PDFGustavo CabanasAún no hay calificaciones
- 4762376Documento54 páginas4762376DrGasnasAún no hay calificaciones
- What Imaging Teaches Us - КопияDocumento62 páginasWhat Imaging Teaches Us - КопияDrGasnas100% (1)
- Music Listening Enhances Cognitive Recovery and Mood After Middle Cerebral Artery StrokeDocumento11 páginasMusic Listening Enhances Cognitive Recovery and Mood After Middle Cerebral Artery StrokeLAURIEJERIEAún no hay calificaciones
- Nervous SystemDocumento123 páginasNervous SystemDrGasnasAún no hay calificaciones
- Central Nervous SystemDocumento25 páginasCentral Nervous SystemDrGasnasAún no hay calificaciones
- Call For Papers: Neural Degeneration and Plasticity in Communication Disorders: Mechanism, Evaluation, and TreatmentDocumento1 páginaCall For Papers: Neural Degeneration and Plasticity in Communication Disorders: Mechanism, Evaluation, and TreatmentDrGasnasAún no hay calificaciones
- Maladaptive Plasticity and Neuropathic PainDocumento1 páginaMaladaptive Plasticity and Neuropathic PainDrGasnasAún no hay calificaciones
- Nervous SystemDocumento58 páginasNervous SystemDrGasnasAún no hay calificaciones
- Nervous SystemDocumento123 páginasNervous SystemDrGasnasAún no hay calificaciones
- Fernandez-Torre ClinNeuroph12-Nonconvulsive SE Adults Electroclin DifferencesDocumento8 páginasFernandez-Torre ClinNeuroph12-Nonconvulsive SE Adults Electroclin DifferencesDrGasnasAún no hay calificaciones
- Call For Papers: Plasticity of The Gabaergic System in Physiology and DiseaseDocumento1 páginaCall For Papers: Plasticity of The Gabaergic System in Physiology and DiseaseDrGasnasAún no hay calificaciones
- WLK Chapter 006 NeurologicAssessDocumento57 páginasWLK Chapter 006 NeurologicAssessDrGasnasAún no hay calificaciones
- UnconsciousnessDocumento91 páginasUnconsciousnessDrGasnasAún no hay calificaciones
- CNS PNSDocumento163 páginasCNS PNSDrGasnasAún no hay calificaciones
- Nervous System Powerpoint2007 - 1Documento98 páginasNervous System Powerpoint2007 - 1DrGasnasAún no hay calificaciones
- ReflexesDocumento35 páginasReflexesBaleli Zagala100% (1)
- Handout2 Fischer CarbeneDocumento5 páginasHandout2 Fischer CarbeneMuhammad ShimaAún no hay calificaciones
- List of Employees with Start and End DatesDocumento394 páginasList of Employees with Start and End DatesMuhammad Faishal TazakkaAún no hay calificaciones
- NCPDocumento18 páginasNCPChristian Karl B. LlanesAún no hay calificaciones
- 08-05-2021 JR - Super60 ActP (In Coming) Jee-Main WTM-01 Question PaperDocumento14 páginas08-05-2021 JR - Super60 ActP (In Coming) Jee-Main WTM-01 Question Paperpurandar puneetAún no hay calificaciones
- Non Ferrous AlloysDocumento45 páginasNon Ferrous AlloysDeepak NegiAún no hay calificaciones
- Duty Resume ReportDocumento1 páginaDuty Resume ReportaleemuddinAún no hay calificaciones
- PositionsDocumento4 páginasPositionsMixsz LlhAdyAún no hay calificaciones
- Periodic Table of Personality ElementsDocumento1 páginaPeriodic Table of Personality Elementslilian_vera_1Aún no hay calificaciones
- Reliance Tabletop SonicDocumento20 páginasReliance Tabletop SonicbrisaAún no hay calificaciones
- PKL Geri RevDocumento3 páginasPKL Geri RevKurniati NiaAún no hay calificaciones
- Jobaid Investigating Causes PDFDocumento16 páginasJobaid Investigating Causes PDFNina MarianaAún no hay calificaciones
- 5 Ethiopian - National - Healthcare - Quality - and - Safety - Strategy - Final - Draft-July122021Documento86 páginas5 Ethiopian - National - Healthcare - Quality - and - Safety - Strategy - Final - Draft-July122021Kemal MahmoudAún no hay calificaciones
- Inspection and Repair of Aircraft Integral Tanks AND Fuel CellsDocumento222 páginasInspection and Repair of Aircraft Integral Tanks AND Fuel CellsgnanasekarAún no hay calificaciones
- Blessed Are Those Who MournDocumento7 páginasBlessed Are Those Who MournPatrick MabbaguAún no hay calificaciones
- Electro BladeDocumento2 páginasElectro Bladeapi-19808945Aún no hay calificaciones
- Butterfly Valve ConcentricDocumento6 páginasButterfly Valve ConcentricpramodtryAún no hay calificaciones
- Guidance Counseling EssentialsDocumento2 páginasGuidance Counseling EssentialsElizabeth E. FetizaAún no hay calificaciones
- t-47 Residential Real Property Affidavit - 50108 ts95421Documento1 páginat-47 Residential Real Property Affidavit - 50108 ts95421api-209878362Aún no hay calificaciones
- Hydrocele: CausesDocumento7 páginasHydrocele: CauseslanaAún no hay calificaciones
- Quartz Textures in Epithermal VeinsDocumento16 páginasQuartz Textures in Epithermal VeinsAlvaro MadridAún no hay calificaciones
- Comparative Analysis of Mineral Constituents of Ethanol Leaf and SeedDocumento9 páginasComparative Analysis of Mineral Constituents of Ethanol Leaf and SeedKIU PUBLICATION AND EXTENSIONAún no hay calificaciones
- RICKETSDocumento23 páginasRICKETSDewi SofyanaAún no hay calificaciones
- Firemac FM Fire Ducts Provide Fire Resistant VentilationDocumento12 páginasFiremac FM Fire Ducts Provide Fire Resistant Ventilationsiva8784Aún no hay calificaciones
- Police Log January 23, 2016Documento9 páginasPolice Log January 23, 2016MansfieldMAPoliceAún no hay calificaciones
- EDC MS 6.4 System DescriptionDocumento10 páginasEDC MS 6.4 System Descriptionmarsh2002Aún no hay calificaciones
- Higuey, Dom Rep Mdpc/Puj: .Eff.23.MayDocumento5 páginasHiguey, Dom Rep Mdpc/Puj: .Eff.23.MayVanessa Yumayusa0% (1)
- Traceability Summary - Supplies July 2015 - June 2016: PT Multimas Nabati Asahan, Kuala TanjungDocumento4 páginasTraceability Summary - Supplies July 2015 - June 2016: PT Multimas Nabati Asahan, Kuala TanjungAbu KhalidAún no hay calificaciones
- Fire & Gas Design BasisDocumento2 páginasFire & Gas Design BasisAdil MominAún no hay calificaciones
- Teardrop by Lauren KateDocumento47 páginasTeardrop by Lauren KateRandom House Teens88% (16)