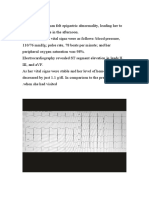
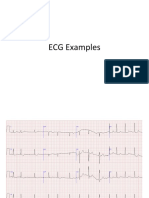
También podría gustarte
- Cric2018 7237454Documento4 páginasCric2018 7237454Muhammad Nur Ardhi LahabuAún no hay calificaciones
- Journal (10 24 22)Documento3 páginasJournal (10 24 22)Louigen DagaydayAún no hay calificaciones
- Selected Topics: Cardiology Commentary: Using Ultrasound To Determine External Pacer CaptureDocumento3 páginasSelected Topics: Cardiology Commentary: Using Ultrasound To Determine External Pacer CaptureJuan Manuel Giraldo SantacolomaAún no hay calificaciones
- Nejmcps 2116690Documento8 páginasNejmcps 2116690Luis MadrigalAún no hay calificaciones
- Stroke 1982 Myers 838 42Documento6 páginasStroke 1982 Myers 838 42Widyawan SyahputraAún no hay calificaciones
- 80 Year Old Patient in Coma After Third StrokeDocumento5 páginas80 Year Old Patient in Coma After Third StrokeRodel Aguila SañoAún no hay calificaciones
- Cardiologia: Hypercalcemic Crisis and Primary Hyperparathyroidism: Cause of An Unusual Electrical StormDocumento5 páginasCardiologia: Hypercalcemic Crisis and Primary Hyperparathyroidism: Cause of An Unusual Electrical StormRo KohnAún no hay calificaciones
- Case Report CardiologiDocumento28 páginasCase Report CardiologiPerdana RichyAún no hay calificaciones
- Anaesthetic Considerations for Mitral Valve ProlapseDocumento7 páginasAnaesthetic Considerations for Mitral Valve ProlapsejaneelsenAún no hay calificaciones
- Department of Internal Medicine II and Physiotherapy Myocarditis CaseDocumento36 páginasDepartment of Internal Medicine II and Physiotherapy Myocarditis CaseSuba Saravanan 12Aún no hay calificaciones
- 22323-Article Text-71358-1-10-20190228 PDFDocumento4 páginas22323-Article Text-71358-1-10-20190228 PDFErna MiraniAún no hay calificaciones
- Overdose and Cardiovascular In: Quinidine Jneurological Toxicity NormalDocumento3 páginasOverdose and Cardiovascular In: Quinidine Jneurological Toxicity NormalNika QuinagonAún no hay calificaciones
- Sudden Cardiac Arrest in Takotsubo Cardiomyopathy - A Case StudyDocumento5 páginasSudden Cardiac Arrest in Takotsubo Cardiomyopathy - A Case StudyzenyxtaAún no hay calificaciones
- Congestive Heart Failure/Pulmonary Edema Case FileDocumento4 páginasCongestive Heart Failure/Pulmonary Edema Case Filehttps://medical-phd.blogspot.comAún no hay calificaciones
- 45 1057Documento5 páginas45 1057Gabriela PinticanAún no hay calificaciones
- Pulmonary Hypertension, Hyperthyroidism, and Fenfluramine: A Case Report and ReviewDocumento42 páginasPulmonary Hypertension, Hyperthyroidism, and Fenfluramine: A Case Report and Reviewthangicu115Aún no hay calificaciones
- Anterolateral Papillary Muscle Rupture As A Result of Single-Vessel Coronary Artery Disease Without Myocardial InfarctionDocumento3 páginasAnterolateral Papillary Muscle Rupture As A Result of Single-Vessel Coronary Artery Disease Without Myocardial InfarctionDonatas AustysAún no hay calificaciones
- Pages From Cardiology - AceQbank MCCQE1 2023 - QbankDocumento10 páginasPages From Cardiology - AceQbank MCCQE1 2023 - QbankDila KavameAún no hay calificaciones
- JURNALDocumento7 páginasJURNALHarlina NurlitaAún no hay calificaciones
- Nej M CPC 1403307Documento8 páginasNej M CPC 1403307Mohamed Saeed El KhayatAún no hay calificaciones
- April 2018 1522659614 97Documento2 páginasApril 2018 1522659614 97Rohit ThakareAún no hay calificaciones
- Acute Coronary SyndromesDocumento7 páginasAcute Coronary SyndromesAiman ArifinAún no hay calificaciones
- Case OneDocumento5 páginasCase Oneola nagarAún no hay calificaciones
- Massive Pulmonary Embolism Presenting As Seizures: December 2008 Vol 10 No 4Documento4 páginasMassive Pulmonary Embolism Presenting As Seizures: December 2008 Vol 10 No 4BelladonnaRoxAún no hay calificaciones
- Rheumatic Heart Disease With Complication of Atrial FibrillationDocumento3 páginasRheumatic Heart Disease With Complication of Atrial FibrillationsamudraandiAún no hay calificaciones
- Myocarditis Due To Systemic Lupus Erythematosus Associated With Cardiogenic ShockDocumento3 páginasMyocarditis Due To Systemic Lupus Erythematosus Associated With Cardiogenic ShockAkbar IskandarAún no hay calificaciones
- Cardiac Sarcoidosis Resembling Panic Disorder: A Case ReportDocumento5 páginasCardiac Sarcoidosis Resembling Panic Disorder: A Case ReportGiiszs AlvarezAún no hay calificaciones
- Review Article: Review and Outcome of Prolonged Cardiopulmonary ResuscitationDocumento9 páginasReview Article: Review and Outcome of Prolonged Cardiopulmonary Resuscitation28121998Aún no hay calificaciones
- Ajmcr 6 8 3Documento4 páginasAjmcr 6 8 3Dewa Aix61Aún no hay calificaciones
- An Unusual, Reversible Cause of Acute High-Outpout HFDocumento5 páginasAn Unusual, Reversible Cause of Acute High-Outpout HFPeter Albeiro Falla CortesAún no hay calificaciones
- Case Report Cteph EditDocumento13 páginasCase Report Cteph EditapekzzzzAún no hay calificaciones
- Electrocardiographic Changes and Intracranial Pathology: Geraldine Syverud, Crna, BSNDocumento4 páginasElectrocardiographic Changes and Intracranial Pathology: Geraldine Syverud, Crna, BSNchuck55Aún no hay calificaciones
- Takotsubo Cardiomyopathy: A Case Series From A Cardiac Center in Rajasthan and Review of LiteratureDocumento8 páginasTakotsubo Cardiomyopathy: A Case Series From A Cardiac Center in Rajasthan and Review of LiteratureIJAR JOURNALAún no hay calificaciones
- Lapkas VT RevisiDocumento13 páginasLapkas VT RevisiRannyAún no hay calificaciones
- Dengue Perimyocarditis: A Case Report: PL GohDocumento3 páginasDengue Perimyocarditis: A Case Report: PL GohDenny IntanAún no hay calificaciones
- DIAGNOSA KERJA DAN DIAGNOSA BANDING Syok KardiogenikDocumento4 páginasDIAGNOSA KERJA DAN DIAGNOSA BANDING Syok KardiogenikPiolitta CyreniaAún no hay calificaciones
- When Past Is Prologue: Clinical Problem-SolvingDocumento7 páginasWhen Past Is Prologue: Clinical Problem-SolvingJoni WitziAún no hay calificaciones
- Electrocardiographic Manifestations of Hypothermia: Amal Mattu, MD, William J. Brady, MD, and Andrew D. Perron, MDDocumento13 páginasElectrocardiographic Manifestations of Hypothermia: Amal Mattu, MD, William J. Brady, MD, and Andrew D. Perron, MDMarta FenandezAún no hay calificaciones
- A Case of Profound Hypothyroidism Presenting With Hypertensive Emergency and Large Amount of Pericardial EffusionDocumento7 páginasA Case of Profound Hypothyroidism Presenting With Hypertensive Emergency and Large Amount of Pericardial EffusionShannya PuaAún no hay calificaciones
- Survival of A Neurologically Intact Patient With Subarachnoid Hemorrhage and Cardiopulmonary ArrestDocumento4 páginasSurvival of A Neurologically Intact Patient With Subarachnoid Hemorrhage and Cardiopulmonary ArrestDino RatnamAún no hay calificaciones
- Pituitary Adenomas Complicating Cardiac Surgery Summary and Review of 11 CasesDocumento8 páginasPituitary Adenomas Complicating Cardiac Surgery Summary and Review of 11 CasessekahaAún no hay calificaciones
- Infective EndocarditisDocumento5 páginasInfective Endocarditismustafa malvi100% (1)
- Criem2020 4159526Documento5 páginasCriem2020 4159526m.fahimsharifiAún no hay calificaciones
- Muerte Subita en DMDocumento10 páginasMuerte Subita en DMChristian Morejon QuezadaAún no hay calificaciones
- Caso Clinico Clinica Mayo 2Documento5 páginasCaso Clinico Clinica Mayo 2Francisco HernandezAún no hay calificaciones
- Case Study on Myocardial Infarction and Heart FailureDocumento33 páginasCase Study on Myocardial Infarction and Heart FailureKelly YeowAún no hay calificaciones
- Assignment: Applied MedicineDocumento15 páginasAssignment: Applied MedicineKhadija BakhtawarAún no hay calificaciones
- ACS AMI FacilitatorDocumento21 páginasACS AMI FacilitatorPaul Zantua57% (7)
- Ischemic Heart Disease: Dr. Amin ShneifiDocumento65 páginasIschemic Heart Disease: Dr. Amin Shneificarmitjulya228Aún no hay calificaciones
- Art2 Eq6 PDFDocumento6 páginasArt2 Eq6 PDFmaky yrigoyenAún no hay calificaciones
- Conservative Management for Anterior STEMI Complicated by Ventricular Septal RuptureDocumento4 páginasConservative Management for Anterior STEMI Complicated by Ventricular Septal RuptureBagus CarterAún no hay calificaciones
- Acute PericarditisDocumento7 páginasAcute PericarditisMirza AlfiansyahAún no hay calificaciones
- Bisoprolol For Fibrilasi AtriumDocumento10 páginasBisoprolol For Fibrilasi AtriumDyanRatihKdeviAún no hay calificaciones
- General Pathology Q2 Heart ConditionsDocumento9 páginasGeneral Pathology Q2 Heart ConditionsAdrian CaballesAún no hay calificaciones
- Clin Cases RF GR 67-15506Documento4 páginasClin Cases RF GR 67-15506simina intunericAún no hay calificaciones
- Case Wecoc FinalDocumento4 páginasCase Wecoc Finalfatmaasri112Aún no hay calificaciones
- Assignment: Applied MedicineDocumento13 páginasAssignment: Applied MedicineKhadija BakhtawarAún no hay calificaciones
- Case-Based Device Therapy for Heart FailureDe EverandCase-Based Device Therapy for Heart FailureUlrika Birgersdotter-GreenAún no hay calificaciones
- Dysrhythmias Nursing InterventionsDocumento7 páginasDysrhythmias Nursing InterventionsGail Lian Santos100% (1)
- Osce PDFDocumento51 páginasOsce PDFAlfonso PeñarroyaAún no hay calificaciones
- Palpitations: DR Polamuri Tabitha PG First YrDocumento37 páginasPalpitations: DR Polamuri Tabitha PG First YrNinaAún no hay calificaciones
- Exam1 100731100921 Phpapp02Documento34 páginasExam1 100731100921 Phpapp02Yaj CruzadaAún no hay calificaciones
- Backwell's 5 Min Veterinary ConsultDocumento120 páginasBackwell's 5 Min Veterinary ConsultAndra Elena Pricop100% (2)
- EKG Interpretation: UNC Emergency Medicine Medical Student Lecture SeriesDocumento58 páginasEKG Interpretation: UNC Emergency Medicine Medical Student Lecture SeriesWidya Surya AvantiAún no hay calificaciones
- Arrhythmia For NursesDocumento49 páginasArrhythmia For NursesRajesh T EapenAún no hay calificaciones
- Electrolyte ImbalanceDocumento4 páginasElectrolyte ImbalanceBharathbushan V MandiriAún no hay calificaciones
- Monitor F. Vitales Philips MP40-50 IntelliVueDocumento390 páginasMonitor F. Vitales Philips MP40-50 IntelliVuechejo84100% (1)
- Lanoxin: (Digoxin) InjectionDocumento35 páginasLanoxin: (Digoxin) InjectionZainAún no hay calificaciones
- DefibrillationDocumento9 páginasDefibrillationJara Maris Moreno BudionganAún no hay calificaciones
- Abel Wakai Diagnosis and Management of Patients WithDocumento2 páginasAbel Wakai Diagnosis and Management of Patients WithJuliaAún no hay calificaciones
- Ecg SimulatorDocumento47 páginasEcg SimulatorSafwan ShaikhAún no hay calificaciones
- Management of Perioperative ArrhythmiasDocumento15 páginasManagement of Perioperative Arrhythmiaszuraini_mdnoorAún no hay calificaciones
- Sudden Cardiac Death in Young AthletesDocumento6 páginasSudden Cardiac Death in Young AthletesnulintavaAún no hay calificaciones
- Practice StripsDocumento9 páginasPractice StripsErica Yamamoto50% (4)
- Obtaining and Interpreting EcgDocumento8 páginasObtaining and Interpreting EcgAndra Elena PricopAún no hay calificaciones
- Taquiarritmias Supraventriculares NEJMDocumento11 páginasTaquiarritmias Supraventriculares NEJMCamilo VidalAún no hay calificaciones
- Supplementary Material - ECG TestDocumento26 páginasSupplementary Material - ECG TestSiraj AL sharifAún no hay calificaciones
- Ecg Short AnswerDocumento3 páginasEcg Short AnswerZoey SanAún no hay calificaciones
- Ecg TestDocumento67 páginasEcg TestEm KayAún no hay calificaciones
- DR Bagus Ari - Cardiogenic Shock KuliahDocumento28 páginasDR Bagus Ari - Cardiogenic Shock KuliahrathalosredAún no hay calificaciones
- Clinical Scenarios in ICUDocumento107 páginasClinical Scenarios in ICUmatenten100% (4)
- LI NarvinDocumento14 páginasLI NarvinRosa Nurul FajriAún no hay calificaciones
- Basic ECG Interpretation - LeonardDocumento33 páginasBasic ECG Interpretation - LeonardAyen FornollesAún no hay calificaciones
- Diagnostic DefibrillationDocumento1 páginaDiagnostic DefibrillationRobAún no hay calificaciones
- Patofisiologi AritmiaDocumento27 páginasPatofisiologi AritmiaVedora Angelia GultomAún no hay calificaciones
- Normal Varient ECGDocumento64 páginasNormal Varient ECGShah NahidAún no hay calificaciones
- ArrhythmiaDocumento31 páginasArrhythmiaAbdallah Essam Al-ZireeniAún no hay calificaciones