También podría gustarte
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeCalificación: 4 de 5 estrellas4/5 (5794)
- X. Li McGue Gottesman 2011 BG-1Documento10 páginasX. Li McGue Gottesman 2011 BG-1FirdousHyderAún no hay calificaciones
- The Little Book of Hygge: Danish Secrets to Happy LivingDe EverandThe Little Book of Hygge: Danish Secrets to Happy LivingCalificación: 3.5 de 5 estrellas3.5/5 (399)
- Plant Extracts For in Vitro in Vivoscreening and Their CharacterizationDocumento34 páginasPlant Extracts For in Vitro in Vivoscreening and Their CharacterizationFirdousHyderAún no hay calificaciones
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryCalificación: 3.5 de 5 estrellas3.5/5 (231)
- Tail Suspension TestDocumento11 páginasTail Suspension TestFirdousHyderAún no hay calificaciones
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceCalificación: 4 de 5 estrellas4/5 (894)
- Writing ProposalsDocumento52 páginasWriting ProposalsRanya MohammedAún no hay calificaciones
- The Yellow House: A Memoir (2019 National Book Award Winner)De EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Calificación: 4 de 5 estrellas4/5 (98)
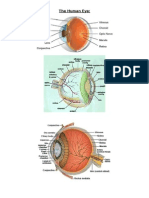
- The Human Eye: Anatomy and FunctionDocumento7 páginasThe Human Eye: Anatomy and FunctionFirdousHyderAún no hay calificaciones
- Shoe Dog: A Memoir by the Creator of NikeDe EverandShoe Dog: A Memoir by the Creator of NikeCalificación: 4.5 de 5 estrellas4.5/5 (537)
- Monthly Current Affairs Quiz - January 2023: Follow UsDocumento244 páginasMonthly Current Affairs Quiz - January 2023: Follow UsSubhankar BasakAún no hay calificaciones
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureCalificación: 4.5 de 5 estrellas4.5/5 (474)
- Ethical Decision Making in ResearchDocumento2 páginasEthical Decision Making in ResearchChandraKurniawanAún no hay calificaciones
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe EverandNever Split the Difference: Negotiating As If Your Life Depended On ItCalificación: 4.5 de 5 estrellas4.5/5 (838)
- Challenges and Opportunities in Preventive and Social MedicineDocumento3 páginasChallenges and Opportunities in Preventive and Social MedicineIJAR JOURNALAún no hay calificaciones
- Grit: The Power of Passion and PerseveranceDe EverandGrit: The Power of Passion and PerseveranceCalificación: 4 de 5 estrellas4/5 (587)
- Attachment Theory and FathersDocumento16 páginasAttachment Theory and FathersGraceAún no hay calificaciones
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaCalificación: 4.5 de 5 estrellas4.5/5 (265)
- Rajiv Gandhi University of Health Sciences Bangalore, Karnataka. Annexure IiDocumento7 páginasRajiv Gandhi University of Health Sciences Bangalore, Karnataka. Annexure IiGirish SubashAún no hay calificaciones
- Week 1 Day 1Documento2 páginasWeek 1 Day 1arens100% (1)
- Pq-Unocal Csms '03Documento15 páginasPq-Unocal Csms '03Ismail Hamzah Azmatkhan Al-husainiAún no hay calificaciones
- The Emperor of All Maladies: A Biography of CancerDe EverandThe Emperor of All Maladies: A Biography of CancerCalificación: 4.5 de 5 estrellas4.5/5 (271)
- LearnEnglish Reading B2 A Plastic Ocean A Film ReviewDocumento5 páginasLearnEnglish Reading B2 A Plastic Ocean A Film ReviewSerly FimasariAún no hay calificaciones
- On Fire: The (Burning) Case for a Green New DealDe EverandOn Fire: The (Burning) Case for a Green New DealCalificación: 4 de 5 estrellas4/5 (73)
- Human Resources Manager or Human Resources Director or Sr. HR BuDocumento2 páginasHuman Resources Manager or Human Resources Director or Sr. HR Buapi-77982767Aún no hay calificaciones
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersCalificación: 4.5 de 5 estrellas4.5/5 (344)
- Child Development A Cultural Approach 2nd Edition Arnett Solutions Manual DownloadDocumento37 páginasChild Development A Cultural Approach 2nd Edition Arnett Solutions Manual DownloadMichael Pontius100% (24)
- Team of Rivals: The Political Genius of Abraham LincolnDe EverandTeam of Rivals: The Political Genius of Abraham LincolnCalificación: 4.5 de 5 estrellas4.5/5 (234)
- Review Article On Chemical Constituents and Uses of Turmeric PlantDocumento8 páginasReview Article On Chemical Constituents and Uses of Turmeric PlantEditor IJTSRDAún no hay calificaciones
- SWP-23 Maintenance Machinery Regime DaimanDocumento1 páginaSWP-23 Maintenance Machinery Regime DaimanHassan AbdullahAún no hay calificaciones
- Human Kinetics Library Platform - Benefits and Values of Outdoor RecreationDocumento17 páginasHuman Kinetics Library Platform - Benefits and Values of Outdoor RecreationMihail RonnyAún no hay calificaciones
- Rise of ISIS: A Threat We Can't IgnoreDe EverandRise of ISIS: A Threat We Can't IgnoreCalificación: 3.5 de 5 estrellas3.5/5 (137)
- Cancer Fighting StrategiesDocumento167 páginasCancer Fighting StrategiesCaptainjillAún no hay calificaciones
- Vulvovaginal Atrophy in The CRETA Study The Healthcare Professionals PerceptionDocumento7 páginasVulvovaginal Atrophy in The CRETA Study The Healthcare Professionals PerceptionHugo GutiérrezAún no hay calificaciones
- The Unwinding: An Inner History of the New AmericaDe EverandThe Unwinding: An Inner History of the New AmericaCalificación: 4 de 5 estrellas4/5 (45)
- MedEx FAQDocumento2 páginasMedEx FAQJee CHIUN CHENAún no hay calificaciones
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyCalificación: 3.5 de 5 estrellas3.5/5 (2219)
- Jordan Belliveau DocumentsDocumento2 páginasJordan Belliveau DocumentsLeigh Egan100% (1)
- SJCTs Pilot Examination Information & InstructionsDocumento6 páginasSJCTs Pilot Examination Information & InstructionsSkep100% (1)
- Elanco Parvovirus DXTX GDocumento2 páginasElanco Parvovirus DXTX Gazamkhan60Aún no hay calificaciones
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreCalificación: 4 de 5 estrellas4/5 (1090)
- Acute Diverticulitis-GcpDocumento85 páginasAcute Diverticulitis-Gcpkuro hanabusaAún no hay calificaciones
- Benefits of Effective Lifting ProgramDocumento30 páginasBenefits of Effective Lifting ProgramMoradeke OnasanyaAún no hay calificaciones
- Non-Steroid Analgesic, Antipyretic, Anti-Inflammatory and Anti-Allergy DrugsDocumento217 páginasNon-Steroid Analgesic, Antipyretic, Anti-Inflammatory and Anti-Allergy DrugsAstri DesmayantiAún no hay calificaciones
- Generic PfPan Job AidDocumento1 páginaGeneric PfPan Job AidSilvia SatoAún no hay calificaciones
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Calificación: 4.5 de 5 estrellas4.5/5 (119)
- Tata AIA Life Insurance Fortune Guarantee Plus Savings PlanDocumento13 páginasTata AIA Life Insurance Fortune Guarantee Plus Savings PlanAkshay SaxenaAún no hay calificaciones
- PALLIATIVE CARE SYMPTOM MANAGEMENTDocumento153 páginasPALLIATIVE CARE SYMPTOM MANAGEMENTrlinao100% (3)
- Patella FractureDocumento52 páginasPatella FractureM Jalil khanAún no hay calificaciones
- Daftar PustakaDocumento3 páginasDaftar PustakaErliTa TyarLieAún no hay calificaciones
- Igem ListDocumento9 páginasIgem ListMilad YadollahiAún no hay calificaciones
- The Perks of Being a WallflowerDe EverandThe Perks of Being a WallflowerCalificación: 4.5 de 5 estrellas4.5/5 (2099)
- High Yield Surgery Compatible VersionDocumento77 páginasHigh Yield Surgery Compatible Version17kimpAún no hay calificaciones
- Serial number verification formDocumento1 páginaSerial number verification formArjelyAún no hay calificaciones
- Her Body and Other Parties: StoriesDe EverandHer Body and Other Parties: StoriesCalificación: 4 de 5 estrellas4/5 (821)