También podría gustarte
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeCalificación: 4 de 5 estrellas4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingDe EverandThe Little Book of Hygge: Danish Secrets to Happy LivingCalificación: 3.5 de 5 estrellas3.5/5 (399)
- Microstructure Effect On Fracture of Stainless SteelsDocumento7 páginasMicrostructure Effect On Fracture of Stainless SteelsAnonymous t7MdBjnOAún no hay calificaciones
- Thesis P Gonzalez RodriguezDocumento165 páginasThesis P Gonzalez RodriguezAnonymous t7MdBjnOAún no hay calificaciones
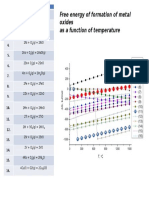
- Free Energy of Formation of Metal Oxides As A Function of TemperatureDocumento1 páginaFree Energy of Formation of Metal Oxides As A Function of TemperatureAnonymous t7MdBjnOAún no hay calificaciones
- Haarman 2016 SRI-IndiaDocumento68 páginasHaarman 2016 SRI-IndiaAnonymous t7MdBjnOAún no hay calificaciones
- The Flow of Real FluidsDocumento35 páginasThe Flow of Real Fluidsbakhtyar21Aún no hay calificaciones
- Art:10.1007/s12289 010 0999 2Documento10 páginasArt:10.1007/s12289 010 0999 2Anonymous t7MdBjnOAún no hay calificaciones
- Lectrure Thermal AnalysisDocumento36 páginasLectrure Thermal Analysisaanfebrianto1993Aún no hay calificaciones
- Coelho e PinhoDocumento12 páginasCoelho e PinhoFrancisco Jesus Patiño MedranoAún no hay calificaciones
- 53 MugeshDocumento5 páginas53 MugeshAnonymous t7MdBjnOAún no hay calificaciones
- Lectrure Thermal AnalysisDocumento36 páginasLectrure Thermal Analysisaanfebrianto1993Aún no hay calificaciones
- Acta Mater 2011 Dual Phase Steel GND Simulation PDFDocumento8 páginasActa Mater 2011 Dual Phase Steel GND Simulation PDFAnonymous t7MdBjnOAún no hay calificaciones
- Secondary Phases Precipitation in DuplexDocumento7 páginasSecondary Phases Precipitation in DuplexAnonymous t7MdBjnOAún no hay calificaciones
- 1 s2.0 S0007850608001947 Main PDFDocumento20 páginas1 s2.0 S0007850608001947 Main PDFOssama RamyAún no hay calificaciones
- 1 - Ecap Valiev Jom 2006 VaznoDocumento7 páginas1 - Ecap Valiev Jom 2006 VaznoAnonymous t7MdBjnOAún no hay calificaciones
- 1 s2.0 S0032386112003254 MainDocumento10 páginas1 s2.0 S0032386112003254 MainAnonymous t7MdBjnOAún no hay calificaciones
- Ltu Ex 2015 103212649Documento112 páginasLtu Ex 2015 103212649Anonymous t7MdBjnOAún no hay calificaciones
- Tribology of Tungsten Disulfide Films in Humid Environments: The Role of A Tailored Metal-Matrix Composite SubstrateDocumento11 páginasTribology of Tungsten Disulfide Films in Humid Environments: The Role of A Tailored Metal-Matrix Composite SubstrateAnonymous t7MdBjnOAún no hay calificaciones
- 10 - Y Saito Acta 1999 PDFDocumento5 páginas10 - Y Saito Acta 1999 PDFAnonymous t7MdBjnOAún no hay calificaciones
- Spectrophotometric Determination of The Copper IonDocumento2 páginasSpectrophotometric Determination of The Copper IonAnonymous t7MdBjnOAún no hay calificaciones
- Structures of Solids & X - Ray DiffractionDocumento14 páginasStructures of Solids & X - Ray DiffractionAnonymous t7MdBjnOAún no hay calificaciones
- WS2 09Jnn LeeDocumento6 páginasWS2 09Jnn LeeAnonymous t7MdBjnOAún no hay calificaciones
- 1 s2.0 S0021929015004406 MainDocumento8 páginas1 s2.0 S0021929015004406 MainAnonymous t7MdBjnOAún no hay calificaciones
- Flexible Conductive Graphene Paper Obtained PDFDocumento36 páginasFlexible Conductive Graphene Paper Obtained PDFAnonymous t7MdBjnOAún no hay calificaciones
- Electrical Communication 45Documento8 páginasElectrical Communication 45Anonymous t7MdBjnOAún no hay calificaciones
- Flexible Conductive Graphene Paper Obtained PDFDocumento36 páginasFlexible Conductive Graphene Paper Obtained PDFAnonymous t7MdBjnOAún no hay calificaciones
- Flexible Conductive Graphene Paper Obtained PDFDocumento36 páginasFlexible Conductive Graphene Paper Obtained PDFAnonymous t7MdBjnOAún no hay calificaciones
- 0912 F 50 FB 2 Ced 2 C 5 Ab 000000Documento10 páginas0912 F 50 FB 2 Ced 2 C 5 Ab 000000Anonymous t7MdBjnOAún no hay calificaciones
- Flexible Conductive Graphene Paper Obtained PDFDocumento36 páginasFlexible Conductive Graphene Paper Obtained PDFAnonymous t7MdBjnOAún no hay calificaciones
- Flexible Conductive Graphene Paper Obtained PDFDocumento36 páginasFlexible Conductive Graphene Paper Obtained PDFAnonymous t7MdBjnOAún no hay calificaciones
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryCalificación: 3.5 de 5 estrellas3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceCalificación: 4 de 5 estrellas4/5 (894)
- The Yellow House: A Memoir (2019 National Book Award Winner)De EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Calificación: 4 de 5 estrellas4/5 (98)
- Shoe Dog: A Memoir by the Creator of NikeDe EverandShoe Dog: A Memoir by the Creator of NikeCalificación: 4.5 de 5 estrellas4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureCalificación: 4.5 de 5 estrellas4.5/5 (474)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe EverandNever Split the Difference: Negotiating As If Your Life Depended On ItCalificación: 4.5 de 5 estrellas4.5/5 (838)
- Grit: The Power of Passion and PerseveranceDe EverandGrit: The Power of Passion and PerseveranceCalificación: 4 de 5 estrellas4/5 (587)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaCalificación: 4.5 de 5 estrellas4.5/5 (265)
- The Emperor of All Maladies: A Biography of CancerDe EverandThe Emperor of All Maladies: A Biography of CancerCalificación: 4.5 de 5 estrellas4.5/5 (271)
- On Fire: The (Burning) Case for a Green New DealDe EverandOn Fire: The (Burning) Case for a Green New DealCalificación: 4 de 5 estrellas4/5 (73)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersCalificación: 4.5 de 5 estrellas4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnDe EverandTeam of Rivals: The Political Genius of Abraham LincolnCalificación: 4.5 de 5 estrellas4.5/5 (234)
- Rise of ISIS: A Threat We Can't IgnoreDe EverandRise of ISIS: A Threat We Can't IgnoreCalificación: 3.5 de 5 estrellas3.5/5 (137)
- The Unwinding: An Inner History of the New AmericaDe EverandThe Unwinding: An Inner History of the New AmericaCalificación: 4 de 5 estrellas4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyCalificación: 3.5 de 5 estrellas3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreCalificación: 4 de 5 estrellas4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Calificación: 4.5 de 5 estrellas4.5/5 (119)
- The Perks of Being a WallflowerDe EverandThe Perks of Being a WallflowerCalificación: 4.5 de 5 estrellas4.5/5 (2099)
- Her Body and Other Parties: StoriesDe EverandHer Body and Other Parties: StoriesCalificación: 4 de 5 estrellas4/5 (821)
- Forced Convection Heat TransferDocumento88 páginasForced Convection Heat TransferSyed YousufuddinAún no hay calificaciones
- Wang 2005Documento6 páginasWang 2005Mohsen MohammadAún no hay calificaciones
- Introduction To Energy PowerpointDocumento24 páginasIntroduction To Energy PowerpointAgha Zeeshan Khan Soomro100% (1)
- Index of Refraction: OutlineDocumento22 páginasIndex of Refraction: OutlineaqsaehsanAún no hay calificaciones
- Aits 1819 FT Vii JeemDocumento23 páginasAits 1819 FT Vii JeemSHREYON SITAMAún no hay calificaciones
- Urea Synthesis With Pool CondenserDocumento1 páginaUrea Synthesis With Pool Condensersite commissing teamAún no hay calificaciones
- 1 AlkalinityDocumento1 página1 AlkalinityprakashAún no hay calificaciones
- Electron ConfigurationDocumento30 páginasElectron ConfigurationShiela Dianne Caliwanagan100% (1)
- Infrared Spectroscopy of Minerals and Related Compounds: Nikita V. Chukanov Alexandr D. ChervonnyiDocumento1116 páginasInfrared Spectroscopy of Minerals and Related Compounds: Nikita V. Chukanov Alexandr D. Chervonnyipellumb shytiAún no hay calificaciones
- Begg Cousland Envirotec - Oil & Gas - 2017Documento8 páginasBegg Cousland Envirotec - Oil & Gas - 2017Wili Nur RahmanAún no hay calificaciones
- Chemistry Jan2012 Unit-1 QPDocumento24 páginasChemistry Jan2012 Unit-1 QPAkila RahmanAún no hay calificaciones
- Textile Chemical Brochure 8.6.22 - 031Documento1 páginaTextile Chemical Brochure 8.6.22 - 031NIKESH PRAKASHAún no hay calificaciones
- Determination of Peracetic Acid by Potentiometric Titration: Sample Preparation and ProceduresDocumento6 páginasDetermination of Peracetic Acid by Potentiometric Titration: Sample Preparation and ProceduresHasan Zeki BayrakAún no hay calificaciones
- Ammonia Mass BalanceDocumento34 páginasAmmonia Mass Balanceaskaridumbo82% (17)
- Steam and Gas TurbinesDocumento2 páginasSteam and Gas TurbinesPunit ShindeAún no hay calificaciones
- Technical Science Notes Students VersionDocumento22 páginasTechnical Science Notes Students VersionAriff AziziAún no hay calificaciones
- 1412 3632Documento8 páginas1412 3632HomeAún no hay calificaciones
- AP Chemistry, Chapter 19, ThermodynamicsDocumento5 páginasAP Chemistry, Chapter 19, Thermodynamicssethisodd100% (2)
- Entropy, Gibbs EnergyDocumento4 páginasEntropy, Gibbs Energyaneece786Aún no hay calificaciones
- COMBUSTIÓN CALCULATIONS USING THE MILLION BTU (1.055 MJ) METHODDocumento9 páginasCOMBUSTIÓN CALCULATIONS USING THE MILLION BTU (1.055 MJ) METHODLuciana RequejoAún no hay calificaciones
- Chemical BondingDocumento44 páginasChemical Bondingjas_ong_man_ling1996Aún no hay calificaciones
- Tasker-Milward School 1Documento19 páginasTasker-Milward School 1Younes AlahmadAún no hay calificaciones
- R134a TableDocumento5 páginasR134a TableAhmed Mahmoud AbouzaidAún no hay calificaciones
- Magneto-Convective Non-Newtonian Nanofluid With Momentum and Temperature Dependent Slip Flow From A Permeable Stretching Sheet With Porous Medium and Chemical ReactionDocumento18 páginasMagneto-Convective Non-Newtonian Nanofluid With Momentum and Temperature Dependent Slip Flow From A Permeable Stretching Sheet With Porous Medium and Chemical ReactionIOSRjournalAún no hay calificaciones
- AttachmentDocumento71 páginasAttachmentAhmad ibrahimAún no hay calificaciones
- Genchem Act#2 ManiulitDocumento3 páginasGenchem Act#2 ManiulitMelissa Kayla ManiulitAún no hay calificaciones
- Electrometallurgy Exam QuestionsDocumento2 páginasElectrometallurgy Exam QuestionsRohan SinghAún no hay calificaciones
- Article 7 Use of Milestones and Constraints1Documento104 páginasArticle 7 Use of Milestones and Constraints1dreamboy87Aún no hay calificaciones
- Axcelis OxygenfreeplasmachipscalepkgDocumento8 páginasAxcelis Oxygenfreeplasmachipscalepkgparam_i47Aún no hay calificaciones
- Computer Simulation of Vapor-Liquid Equilibrium in Mixed Solvent Electrolyte SolutionsDocumento10 páginasComputer Simulation of Vapor-Liquid Equilibrium in Mixed Solvent Electrolyte SolutionsJoseCastilhoAún no hay calificaciones